INTRODUCTION GÉNÉRALE À l 'IDRIS
Ce document s'adresse principalement aux nouveaux utilisateurs de l'IDRIS. Il présente les principaux points indispensables à une bonne utilisation des machines de l'IDRIS.
1. Présentation de l'IDRIS
2. Présentation de la machine
3. Demandes d'allocations d'heures sur la machine de l'IDRIS
4. Comment obtenir un compte ?
5. Comment se connecter pour calculer ?
6. Gestion de son compte et de son environnement
7. Présentation des espaces disques
8. Commandes de transferts de fichiers
9. La commande module
10. Compilation
11. Exécution
12. Formations proposées à l'IDRIS
13. Documentation de l'IDRIS
14. Le support aux utilisateurs
Pour une documentation complète sur les différents points abordés dans ce document, n’hésitez pas à consulter le site de l'IDRIS : www.idris.fr
1. Présentation de l'IDRIS
Missions et objectifs de l'IDRIS
L’IDRIS (Institut du développement et des ressources en informatique scientifique), fondé en novembre 1993, est le centre national du CNRS pour le calcul numérique intensif de très haute performance (HPC) et l’intelligence artificielle (IA) par apprentissage profond, au service des communautés scientifiques de la recherche publique ou privée (sous condition de recherche ouverte avec publication des résultats), tributaires de l'informatique extrême.
À la fois centre de ressources informatiques et pôle de compétences en HPC et IA, l'IDRIS (www.idris.fr) est une unité d'appui et de recherche du CNRS (UAR 851), dépendant de la Direction des données ouvertes de la recherche (DDOR) du CNRS et rattachée administrativement à l’Institut des Sciences informatiques (INS2I), mais dont la vocation à l’intérieur du CNRS est pluridisciplinaire. Les modalités de fonctionnement de l’IDRIS sont proches de celles des IR* (Infrastructures de Recherche “étoile”) du Ministère de l'Enseignement supérieur et de la Recherche (MESR).
Gestion scientifique des ressources
L'attribution des ressources de calcul de l'ensemble des centres nationaux (CINES, IDRIS et TGCC) est organisée sous la coordination du GENCI (Grand équipement national de calcul intensif).
Les demandes de ressources se font via le portail eDARI pour l'ensemble des centres .
Il est possible de déposer des demandes d’heures de calcul pour un nouveau projet, pour renouveler un projet existant ou pour compléter une allocation obtenue précédemment. Les allocations d'heures attribuées sont valables un an.
Suivant le volume d'heures demandé, le dossier déposé sera de type accès régulier (AR) ou accès dynamique (AD).
Dans le cas des accès réguliers : l'appel à projet pour les accès réguliers est ouvert en permanence mais l'évaluation des dossiers est semestrielle au sein des campagnes d'attribution. Les dossiers sont évalués du point de vue scientifique par les membres des comités thématiques en s'appuyant le cas échéant sur l'expertise technique, réalisé par les équipes d'assistance applicative des centres. Puis un Comité d'évaluation se réunit pour statuer sur les demandes de ressources et indiquer au Comité d'attribution, placé sous la responsabilité de GENCI, les propositions d'affectation des heures de calculs sur les trois centres nationaux.
Dans le cas des accès dynamique : les demandes sont examinées par le directeur de l'IDRIS qui jugera de la qualité scientifique et technique des dossiers et pourra demander l'avis d'un expert du comité thématique si besoin.
Dans les deux cas, la direction de l'IDRIS étudie les demandes spécifiques (dites “au fil de l'eau”) et effectue des attributions limitées pour éviter le blocage de projets en cours.
Pour plus d'information, consulter la page : Demandes d'allocations d'heures sur la machine de l'IDRIS).
Vous trouverez également une vidéo explicative concernant les demandes d'heures et d'ouverture de compte sur Jean Zay sur notre chaîne YouTube "Un œil sur l'IDRIS" :
Le Comité des utilisateurs de l'IDRIS
Le rôle du comité des utilisateurs est de dialoguer avec le centre afin que tous les projets auxquels des ressources informatiques ont été attribuées puissent être menés à bien dans les meilleures conditions. Il transmet les observations de tous les utilisateurs sur le fonctionnement du centre et dialogue avec l'IDRIS pour définir les évolutions souhaitables.
Idéalement, le comité des utilisateurs est constitué de 2 élus par discipline scientifique qui peuvent être contactés collectivement à l'adresse : Les pages du CU sont disponibles pour les utilisateurs de l'IDRIS en se connectant à l'Extranet de l'IDRIS, section Comité des utilisateurs.
C'est dans cet espace que sont mis à disposition les bilans d'exploitation des machines de l'IDRIS ainsi que les compte-rendu des dernières réunions.
Groupe de travail pour des demandes FSD uniques
Un groupe de travail est en place depuis 2 ans environs. Y participent la responsable du service PPST (Protection du Potentiel Scientifique et Technique) du FSD (Fonctionnaire Sécurité Défense), 3 personnes de l’IDRIS dont le directeur et des représentants des utilisateurs :
- Marie-Bernadette.Lepetit@neel.cnrs.fr ; Tel 04.76.88.90.45
- Alain.Miniussi@oca.eu Tel : +33 4 92 00 30 09 / +33 4 83 61 85 44
Les utilisateurs demandent depuis des années qu’il ne soit nécessaire de faire qu'une unique demande d'autorisation FSD pour les 3 centres de calcul nationaux et que la réponse soit valide pour les 3 centres.
Cette demande a été accepté sur le principe depuis longtemps, mais n’est encore que partiellement en place. Ce groupe travaille à ce que cela soit mis en place le plus rapidement possible. Nous invitons toute personne pour qui cela ne fonctionnerait pas (propagation automatique sans avoir à refaire les demandes) à contacter la présidente du Comité des Utilisateurs (Marie-Bernadette.Lepetit@neel.cnrs.fr ; Tel 04.76.88.90.45).
Les membres du Comité Utilisateur participant au groupe de travail vont aussi endosser un rôle de correspondants PPST :
- conseil sur les procédures pour les demandes FSD
- répondre aux questions courantes des utilisateurs
- relai des problèmes rencontrés vis à vis des instances FSD
- relai vers les utilisateurs des informations sur les procédures pour être accepté sur les centres de calcul.
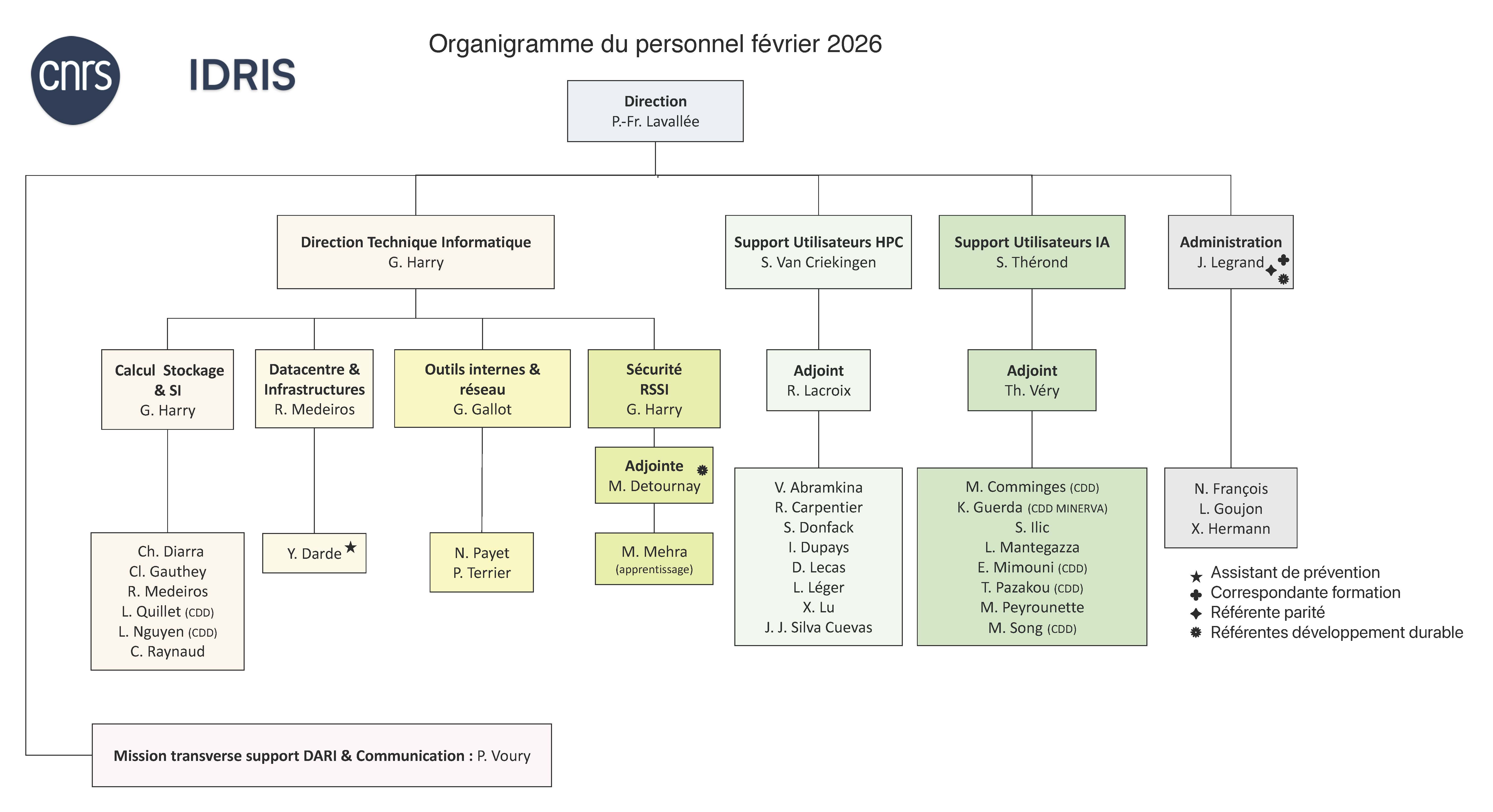
Le personnel de l'IDRIS
2. Présentation de la machine
Jean Zay : calculateur Eviden BullSequana XH3000, HPE SGI 8600
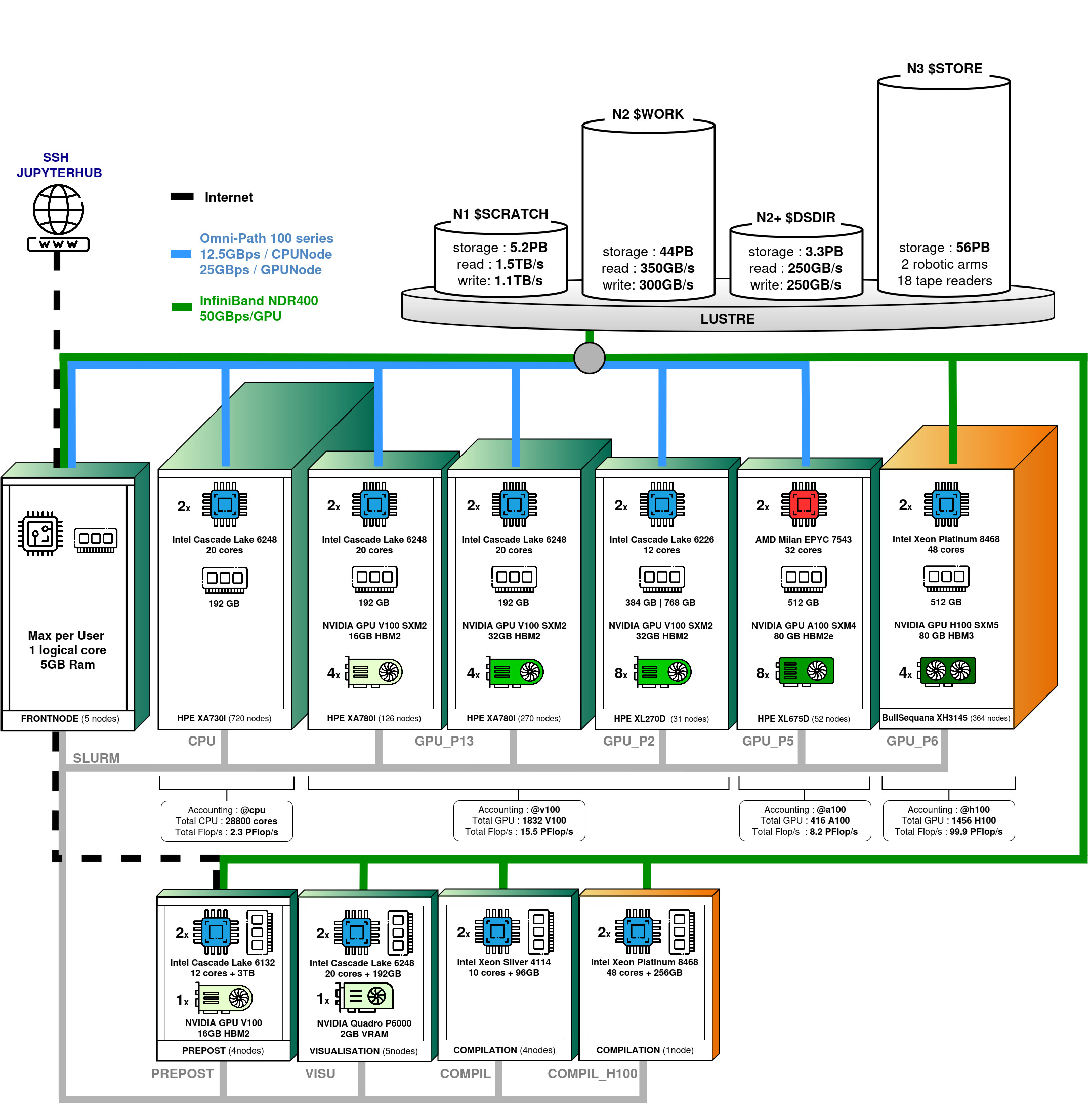
Jean Zay est un supercalculateur comportant un partie Eviden BullSequana XH3000 et une partie HPE SGI 8600 formant un total de cinq partitions : une partition contenant des nœuds scalaires (ayant uniquement des CPU) et quatre partitions contenant des nœuds accélérés (nœuds hybrides équipés à la fois de CPU et de GPU). Les nœuds de calcul HPE SGI 8600 sont interconnectés par un réseau Intel Omni-PAth et ceux de l'extension Eviden BullSequana XH3000 sont interconnectés par un réseau Infiniband. L'ensemble des nœuds accède à un système de fichiers partagé à très forte bande passante.
Après trois extensions successives, la puissance crête cumulée de Jean Zay est, depuis juillet 2024, de 125,9 Pétaflop/s.
L'accès aux diverses partitions hardware de la machine dépend du type de travail soumis (CPU ou GPU) et de la partition Slurm demandée pour son exécution (voir le détail des partitions Slurm CPU et des partitions Slurm GPU).
Partition scalaire (ou partition CPU)
Sans indiquer de partition CPU ou avec la partition cpu_p1, vous aurez accès aux ressources suivantes :
- 720 nœuds de calcul scalaires avec :
- 2 processeurs Intel Xeon Gold 6248 (20 cœurs à 2,5 GHz), soit 40 cœurs par nœud
- 192 Go de mémoire par nœud
Remarque : suite au décommissionnement de 808 nœuds CPU le 05/02/2024, cette partition est passée de 1528 nœuds à 720 nœuds.
Partitions accélérées (ou partitions GPU)
Sans indiquer de partition GPU ou avec la contrainte v100-16g ou v100-32g, vous aurez accès aux ressources suivantes :
- 396 nœuds de calcul accélérés quadri-GPU avec :
- 2 processeurs Intel Xeon Gold 6248 (20 cœurs à 2,5 GHz), soit 40 cœurs par nœud
- 192 Go de mémoire par nœud
- 126 nœuds avec 4 GPU Nvidia Tesla V100 SXM2 16 Go (avec v100-16g)
- 270 nœuds avec 4 GPU Nvidia Tesla V100 SXM2 32 Go (avec v100-32g)
Remarque : suite au décommissionnement de 220 nœuds 4-GPU V100 16 Go (v100-16g) le 05/02/2024, cette partition est passée de 616 nœuds à 396 nœuds.
Avec la partition gpu_p2, gpu_p2s ou gpu_p2l, vous aurez accès aux ressources suivantes :
- 31 nœuds de calcul accélérés octo-GPU avec :
- 2 processeurs Intel Xeon Gold 6226 (12 cœurs à 2,7 GHz), soit 24 cœurs par nœud
- 20 nœuds à 384 Go de mémoire (avec gpu_p2 ou gpu_p2s)
- 11 nœuds à 768 Go de mémoire (avec gpu_p2 ou gpu_p2l)
- 8 GPU Nvidia Tesla V100 SXM2 32Go
Avec la partition gpu_p5 accessible uniquement avec des heures GPU A100 (extension juin 2022), vous aurez accès aux ressources suivantes :
- 52 nœuds de calcul accélérés octo-GPU avec :
- 2 processeurs AMD Milan EPYC 7543 (32 cœurs à 2,80 GHz), soit 64 cœurs par nœud
- 512 Go de mémoire par nœud
- 8 GPU Nvidia A100 SXM4 80 Go
Avec la partition gpu_p6 accessible uniquement avec des heures GPU H100 (extension été 2024), vous aurez accès aux ressources suivantes :
- 364 nœuds de calcul accélérés quadri-GPU avec :
- 2 processeurs Intel Xeon Platinum 8468 (48 cœurs à 2,10 GHz), soit 96 cœurs par nœud
- 512 Go de mémoire par nœud
- 4 GPU Nvidia H100 SXM5 80 Go
Pré et post-traitement
Avec la partition prepost, vous aurez accès aux ressources suivantes :
- 4 nœuds de pré et post-traitement à large mémoire avec :
- 4 processeurs Intel Skylake 6132 (12 cœurs à 3,2 GHz), soit 48 cœurs par nœud
- 3 To de mémoire par nœud
- 1 GPU Nvidia Tesla V100
- un disque interne NVMe de 1,5 To
Visualisation
Avec la partition visu, vous aurez accès aux ressources suivantes :
- 5 nœuds de visualisation de type scalaire
- 2 processeurs Intel Cascade Lake 6248 (20 cœurs à 2,5 GHz), soit 40 cœurs par nœud
- 192 Go de mémoire par nœud
- 1 GPU Nvidia Quatro P6000
Compilation
Avec la partition compil, vous aurez accès aux ressources suivantes :
- 4 nœuds de pré et post-traitement (voir ci-dessus)
- 3 nœuds de compilation
- 1 processeur Intel(R) Xeon(R) Silver 4114 (10 cœurs à 2,20 GHz)
- 96 Go de mémoire par nœud
Une partition compil_h100 est également disponible, donnant accès à un processeur identique à ceux de la partition Eviden H100 (gpu_p6) :
- 1 nœud de compilation
- 1 processeur Intel(R) Xeon(R) Platinum 8468 (48 cœurs à 2,10 GHz)
- 256 Go de mémoire par nœud
Archivage
Avec la partition archive, vous aurez accès aux ressources suivantes :
- 4 nœuds de pré et post-traitement (voir ci-dessus)
Autres caractéristiques
- Puissance crête cumulée : 126 PFlop/s (depuis le 5/02/2024).
- Un réseau d'interconnexion Intel Omni-PAth 100 Gb/s : 1 lien par nœud scalaire et 4 liens par nœud convergé V100 (quadri-GPU ou octo-GPU) et A100 (octo-GPU)
- Un réseau d'interconnexion Infiniband NDR 400 Gb/s : 4 liens par nœud convergé H100 (quadri-GPU)
- Un système de fichiers parallèle Lustre
- Un dispositif de stockage parallèle avec des disques SSD d'une capacité de 2,5 Po (GridScaler GS18K SSD) suite à l'extension de l'été 2020.
- Un dispositif de stockage parallèle avec des disques d'une capacité supérieure à 30 Po
- 5 nœuds frontaux
- 2 processeurs Intel Cascade Lake 6248 (20 cœurs à 2,5 GHz), soit 40 cœurs par nœud
- 192 Go de mémoire par nœud
3. Demandes d'allocations d'heures sur la machine de l'IDRIS
Demandes d'allocations d'heures à l'IDRIS
Les demandes d'allocations d'heures sur Jean Zay se font via le portail eDARI commun aux 3 centres nationaux CINES, IDRIS, et TGCC : www.edari.fr.
Avant toute demande d'heure, nous vous recommandons de consulter la note GENCI détaillant les modalités d'accès aux ressources nationales. Vous y trouverez entre autres, les conditions et critères d’éligibilité pour obtenir des heures de calcul.
Quel que soit l'usage que vous envisagez (IA ou HPC) vous pouvez postuler à tout moment via un formulaire unique sur le portail eDARI, à un Accès Dynamique pour les demandes ≤ 50 kh GPU normalisées (1 heure A100 = 2 heures V100 = 2 heures GPU normalisées) / 500 kh CPU ou à un Accès Régulier pour les demandes plus importantes. Attention : il s'agit de votre demande de ressources CUMULÉE dans les trois centres nationaux. En fonction du volume de ressources demandées, votre dossier sera de type Accès Dynamique ou Accès Régulier
A partir de votre espace personnel (compte eDARI) sur le portail eDARI vous pouvez :
- constituer un dossier d’accès dynamique ou régulier,
- renouveler un dossier d’accès dynamique ou régulier,
- demander l'ouverture d'un compte de calcul nécessaire pour accéder aux ressources de calcul sur Jean Zay. Plus d'information sur la page sur la gestion des comptes de l'IDRIS.
Vous trouverez une vidéo explicative concernant les demandes d'heures et d'ouverture de compte sur Jean Zay sur notre chaîne YouTube "Un œil sur l'IDRIS" :
Accès dynamiques (AD)
Les demandes de ressources de types accès dynamiques sont possibles toute l'année et sont renouvelables. Les demandes sont expertisées et validées par le directeur de l'IDRIS. L'allocation d'heures est valable un an à partir de l'ouverture du compte de calcul sur Jean Zay.
Accès réguliers (AR)
Deux appels à projets pour des accès réguliers sont lancés chaque année :
- en janvier-février pour une attribution des heures du 1er mai de la même année au 30 avril de l'année suivante
- et en juin-juillet pour une attribution des heures du 1er novembre de l’année courante au 31 octobre de l'année suivante.
Les nouveaux dossiers d'accès réguliers peuvent être déposés tout au long de l'année. ils sont expertisés dans le cadre de la campagne dont la date limite de clôture suit immédiatement la validation du dossier par son porteur de projet.
Les renouvellements d’accès réguliers, doivent eux, être déposés dans le cadre des campagnes avant la date de clôture pour une expertise dans le cadre de cette campagne.
Pour information, la date limite de clôture des prochains appels est visible sur le site web du DARI (première colonne à gauche).
Demande au fil de l'eau
Tout au long de l'année, à partir du portail eDARI, vous avez la possibilité de faire des demandes au fil de l'eau pour tous les projets existants (accès dynamiques et accès réguliers HPC) ayant épuisé leurs quotas d'heures initial. Pour les ADs, la demande globale doit rester inférieure au seuil de 50 000 h GPU ou 500 000 h CPU.
Documentations
Deux documentations références sont à votre disposition :
- la documentation IDRIS pour vous aider à réaliser chacune de ces formalités via le portail eDARI.
- la note GENCI détaillant les modalités d'accès aux ressources nationales. La bible.
4. Comment obtenir un compte ?
Gestion des comptes : ouverture et fermeture
Le compte utilisateur
Chaque utilisateur dispose d'un unique compte pouvant être associé à tous les projets auxquels il participe.
Pour plus d'information, vous pouvez consulter notre page web concernant la gestion des comptes multi-projet.
Le formulaire de gestion de compte FGC permet d'effectuer des modifications sur un compte existant : ajout/suppression de points d'accès, changement d'adresse postale, de téléphone, d'employeur, etc…
Un compte a deux états possibles :
- ouvert ; dans ce cas vous pouvez :
- soumettre des travaux sur la machine de calcul si l'allocation du projet n'est pas épuisée (cf. sortie de la commande idracct) ;
- soumettre des travaux de pré/post traitement ;
- fermé ; dans ce cas vous ne pouvez plus vous connecter sur la machine. Une notification par courriel est adressée au responsable du projet et à l'utilisateur lors de la fermeture.
Ouverture du compte d'un utilisateur
Cas d'un nouveau projet
Il n'y a pas d'ouverture de compte automatique ou implicite. Chaque utilisateur d'un projet doit donc demander :
- s'il n'a pas encore de compte IDRIS, l'ouverture d'un compte de calcul respectant les modalités d'accès définies par GENCI. Cela se fait, pour un accès dynamique aussi bien que pour un accès régulier, à partir du portail eDARI.
- si il a déjà un compte ouvert à l'IDRIS, le rattachement de son compte au projet concerné via le portail eDARI.
ATTENTION : sur décision du directeur de l’IDRIS ou du Fonctionnaire de Sécurité et de Défense (FSD) du CNRS, la création d’un nouveau compte peut être soumise à autorisation ministérielle, dans le cadre de l’application de la réglementation de Protection du Potentiel Scientifique et Technique de la nation (PPST). Dans un tel cas, une communication personnelle sera transmise afin de mettre en œuvre la procédure requise, sachant que l’instruction du dossier peut demander jusqu’à deux mois. Pour anticiper cela, vous pouvez envoyer un mail directement au centre concerné (voir les contacts sur le portail eDARI) pour initier la procédure en amont (pour vous-même ou pour quelqu'un que vous souhaitez prochainement intégrer dans votre groupe) avant même que la demande d'ouverture de compte soit envoyée pour validation au responsable de la structure de recherche et au correspondant sécurité de la structure de recherche.
Remarque : L'ouverture effective d'un nouveau compte sur la machine ne sera faite que lorsque la demande d'accès (régulier ou dynamique) est validée (autorisation ministérielle) et que le projet correspondant a obtenu des heures de calcul.
Vous trouverez une vidéo explicative concernant les demandes d'heures et d'ouverture de compte sur Jean Zay sur notre chaîne YouTube "Un œil sur l'IDRIS" :
Cas d'un renouvellement de projet
Les comptes existants sont automatiquement reportés d'un appel à projet à l'autre si les conditions d’éligibilité des membres du projet n'ont pas changé entre-temps (cf. la note GENCI décrivant les modalités d'accès aux ressources GENCI).
Par conséquent, si votre compte est ouvert et déjà associé au projet qui a obtenu des heures pour l'appel suivant aucune démarche n'est nécessaire.
Fermeture d'un compte utilisateur
Fermeture suite au non renouvellement du projet
Lorsqu'un projet GENCI n'est pas renouvelé, la procédure appliquée est la suivante :
- à la date d'expiration de l'allocation d'heures du projet :
- les heures DARI non consommées ne sont plus disponibles. Les comptes du projet ne peuvent plus soumettre de travaux sur la machine de calcul pour ce projet (plus de comptabilité d'heures pour le projet) ;
- les comptes restent ouverts et rattachés au projet pour permettre l'accès aux données du projet pendant 6 mois ;
- six mois après la date d'expiration de l'allocation d'heures :
- Les comptes sont détachés du projet (plus aucun accès aux données du projet) ;
- les données du projet (SCRATCH, STORE, WORK, ALL_CCFRSCRATCH, ALL_CCFRSTORE et ALL_CCFRWORK) seront supprimées à l'initiative de l'IDRIS dans un délai non défini ;
- les comptes encore rattachés à un autre projet restent ouverts mais, si le projet non renouvelé était leur projet par défaut, il doivent le changer via la commande idrproj (sinon les variables (SCRATCH, STORE, WORK, ALL_CCFRSCRATCH, ALL_CCFRSTORE et ALL_CCFRWORK ne seront pas définies) ;
- les comptes qui ne sont plus rattachés à aucun projet peuvent alors être fermés à tout moment.
La récupération des fichiers est à la charge de chaque utilisateur pendant les six mois qui suivent la fin d'un projet non renouvelé, en transférant les fichiers sur une machine locale au laboratoire de l'utilisateur ou sur les espaces disque Jean Zay d'un autre projet DARI pour les comptes multi-projets.
Ces six mois de délai permettent d'éviter de fermer inutilement les comptes d'un projet à la fin de l'allocation Ai si ce dernier n'a pas été renouvelé l'année suivante (pas de demande d'heures pour l'allocation Ai+2) mais si il a été renouvelé pour la période d'allocation Ai+3 (soit 6 mois après Ai+2).
Fermeture suite à l'expiration de l'autorisation ministérielle d'accès aux ressources informatiques de l'IDRIS
Les autorisations ministérielles ne sont accordées/valides que pour une période donnée. Lorsque votre autorisation ministérielle atteint sa date d'expiration, nous sommes dans l'obligation de fermer votre compte.
Dans ce cas, la procédure est la suivante :
- envoi d'une première notification par courriel 90 jours avant la date d'expiration,
- envoi d'une deuxième notification par courriel 70 jours avant la date d'expiration
- le compte est fermé le jour de l'expiration si l'autorisation n'a pas été renouvelée.
Important : pour éviter cette fermeture, dès réception du premier courriel, l’utilisateur est invité à faire une nouvelle demande d'ouverture de compte via le portail eDARI pour que l'IDRIS puisse démarrer l'instruction d'un dossier de prolongation. En effet, l’instruction du dossier peut demander jusqu’à deux mois.
Fermeture pour raison de sécurité
Un compte peut être fermé à tout moment et sans préavis sur décision de la Direction de l'IDRIS.
Fermeture suite à une demande de détachement formulée par le chef de projet
Le chef de projet peut demander le détachement d'un compte rattaché à son projet en complétant et en nous envoyant le formulaire de gestion de compte (FGC).
Lors de cette demande, le chef de projet peut demander que les données du compte détaché et contenues dans les répertoires SCRATCH, STORE, WORK, ALL_CCFRSCRATCH, ALL_CCFRSTORE et ALL_CCFRWORK soient immédiatement supprimées ou recopiées dans les répertoires d'un autre utilisateur rattaché au même projet.
Mais suite au détachement, si le compte détaché n'est plus rattaché à aucun projet, il peut alors être fermé à tout moment.
Déclaration des machines à partir desquelles un utilisateur se connecte à l'IDRIS
Toute machine utilisée pour accéder à un calculateur de l'IDRIS doit être enregistrée dans les filtres de l'IDRIS.
Pour cela, tout utilisateur doit fournir, lors de la demande de création de compte, la liste des machines avec lesquelles il se connectera au calculateur de l'IDRIS (adresse IP et nom). Il fait cette déclaration au moment de la création de compte utilisateur à partir du portail eDARI.
La mise à jour (ajout/suppression) de la liste des machines associées à un login peut être demandée par son propriétaire à l'aide du formulaire FGC (Formulaire de gestion de compte). Ce formulaire doit être complété et signé par l’utilisateur et le responsable sécurité du laboratoire.
Attention : les adresses IP personnelles ne sont pas autorisées pour se connecter aux machines de l'IDRIS.
Responsable sécurité du laboratoire
Le responsable sécurité du laboratoire est l'interlocuteur réseau /sécurité du laboratoire vis à vis de l'IDRIS. Il doit garantir que la configuration de la machine à partir de laquelle l'utilisateur se connecte à l'IDRIS est conforme aux règles et usages les plus récents en matière de sécurité informatique et doit pouvoir fermer immédiatement l'accès de l'utilisateur à l'IDRIS en cas d'alerte de sécurité.
Son nom et ses coordonnées sont transmis à l'IDRIS par le directeur du laboratoire à l'aide du formulaire FGC (Formulaire de gestion de compte). Ce formulaire est aussi utilisé pour informer l'IDRIS en cas de changement de responsable sécurité.
Comment accéder à l'IDRIS en télétravail ou en mission ?
Pour des raisons de sécurité, nous ne pouvons pas autoriser l'accès aux machines de l'IDRIS depuis des adresses IP non institutionnelles. Il est donc par exemple exclu que vous puissiez vous connecter directement depuis votre connexion personnelle.
Utilisation d'un VPN
La solution recommandée pour accéder aux ressources de l'IDRIS lorsque vous êtes en mobilité (télétravail, mission, etc) est d'utiliser le service VPN (Virtual Private Network) de votre laboratoire/institut/université. Un VPN vous permet d'accéder à des ressources distantes comme si vous étiez connecté directement au réseau local de votre laboratoire. Il reste néanmoins nécessaire d'enregistrer l'adresse IP attribuée à votre machine par le VPN en suivant la procédure décrite plus haut.
Cette solution a l'avantage de permettre l'utilisation des services de l'IDRIS accessibles via un navigateur web (par exemple l'extranet ou des produits comme Jupyter Notebook, JupyterLab et TensorBoard)
Utilisation d'une machine de rebond
Si l'utilisation d'un VPN est impossible, il est toujours possible de vous connecter en SSH à une machine de rebond de votre laboratoire, depuis laquelle Jean Zay est accessible (ce qui implique d'avoir fait enregistrer l'adresse IP de cette machine rebond).
vous@ordinateur_portable:~$ ssh login_rebond@machine_rebond login_rebond@machine_rebond:~$ ssh login_idris@machine_idris
Notez qu'il est possible d'automatiser le rebond via les options ProxyJump ou ProxyCommand de SSH pour pouvoir vous connecter en utilisant une seule commande (par exemple ssh -J login_rebond@machine_rebond login_idris@machine_idris).
Comment accéder ponctuellement à l'IDRIS depuis l'étranger ?
La demande d'autorisation de la machine doit être faite par le missionnaire qui complète le 2e cadre page 3 du formulaire FGC (“Cadre à remplir dans le cas d’un séjour à l’étranger”). Un accès ssh temporaire sur l'ensemble des machines du centre est alors activé.
5. Comment se connecter pour calculer ?
Comment accéder à Jean Zay ?
Chaque utilisateur de l'IDRIS est titulaire d'un login unique pour tous les projets auxquels il participe. Ce compte est personnel et ne doit pas être partagé. Ce login est associé à un mot de passe obéissant à certaines règles de sécurité. Avant de vous connecter, nous vous conseillons de consulter la page gestion de mot de passe.
Vous ne pouvez vous connecter sur Jean Zay qu'à partir d'une machine dont l'adresse IP est enregistrée dans nos filtres. Si cela n'est pas le cas, consultez la procédure de déclaration des machines disponible sur notre site Web.
Les accès interactifs à Jean Zay sont possibles via le protocole ssh sur les nœuds frontaux de la machine ou via notre instance JupyterHub (attention, une première connexion via ssh est nécessaire pour définir votre mot de passe).
Pour de plus amples informations, vous pouvez consulter la description matérielle détaillée du cluster.
Jean Zay : accès et shells
Accès aux machines
Jean Zay :
La connexion à la frontale de Jean Zay se fait par ssh depuis une machine enregistrée à l'IDRIS :
$ ssh mon_login_idris@jean-zay.idris.fr
puis saisissez votre mot de passe, si vous n'avez pas configuré de clef ssh.
Jean Zay pré et post-traitement :
La connexion interactive à la frontale de pré et post-traitement se fait par ssh depuis une machine enregistrée à l'IDRIS :
$ ssh mon_login_idris@jean-zay-pp.idris.fr
puis saisissez votre mot de passe, si vous n'avez pas configuré de clef ssh.
Connexion par clef ssh
les connexions SSH utilisant une paire de clefs SSH (clé privée / clé publique) sont possibles sur Jean Zay.
ATTENTION : nous envisageons de renforcer notre politique de sécurité concernant les accès à la machine jean-zay. Par conséquent, nous vous demandons de tester, dès maintenant, l'utilisation des certificats pour vos connexions SSH en lieu et place des paires de clés SSH habituelles (clé privée / clé publique) en respectant les procédures détaillées ici.
Connexion ssh avec certificat
Pour renforcer notre politique de sécurité concernant les accès à la machine jean-zay, nous vous demandons de tester l'utilisation des certificats pour vos connexions SSH en lieu et place des paires de clés SSH habituelles (clé privée / clé publique). La création et l'utilisation de certificats se fait en respectant les procédures détaillées ici.
Pendant la phase de test, les connexions via les clés SSH classiques resteront possibles. Merci de nous signaler tous les problèmes que vous pourriez rencontrer avec l'utilisation des certificats.
Gestion de son environnement
Votre espace $HOME est commun à toutes les frontales de Jean-Zay. Par conséquent, chaque modification de vos fichiers d'environnement personnels s'applique automatiquement sur toutes les machines.
Quels sont les shells disponibles sur les machines de l'IDRIS ?
Le Bourne Again shell (bash) est le seul interpréteur de commandes supporté comme shell de login sur les machines de l'IDRIS : l'IDRIS ne garantit pas que l'environnement utilisateur par défaut soit correctement défini avec les autres shells. Le bash est une évolution importante du Bourne shell (ancien sh) avec des fonctionnalités avancées.
Cependant, d'autres interpréteurs (ksh, tcsh, csh) sont aussi installés sur les machines pour permettre l'exécution de scripts utilisant ces shells.
Quels sont les fichiers d'environnement invoqués lors du lancement d'une session login en bash ?
Le fichier .bash_profile, s'il existe dans votre HOME, est exécuté au login une seule fois pendant une session. Sinon c'est le fichier .profile qui est exécuté, s'il existe. C'est dans un de ces fichiers que l'on place les variables d'environnement, les programmes à lancer à la connexion. La définition des alias, des fonctions personnelles et le chargement de modules sont à mettre dans le fichier .bashrc qui, lui, est exécuté au lancement de chaque sous-shell.
Il est préférable de n'utiliser qu'un seul fichier d'environnement : le .bash_profile ou .profile.
Gestion des mots de passe
La connexion à Jean Zay se fait par la saisie de votre login et du mot de passe associé.
Pour votre première connexion vous devez utiliser un mot de passe initial qui sera changé immédiatement, afin de définir votre mot de passe courant.
Le mot de passe initial
Qu'est ce que le mot de passe initial ?
Le mot de passe initial est le résultat de la concaténation de deux mots de passe dans l'ordre suivant :
- Premièrement, le mot de passe généré aléatoirement par l'IDRIS, qui vous est envoyé par courriel lors de l'ouverture de votre compte et lors de la réinitialisation de votre mot de passe. Il reste valide 20 jours.
- Deuxièmement, le mot de passe initial utilisateur (8 caractères), que vous devez renseigner :
- lors de la demande d'ouverture de votre compte (si vous êtes nouvel utilisateur) sur le portail DARI ;
- lors d'une demande de réinitialisation du mot de passe initial dans le Formulaire de Gestion de Compte (FGC).
Remarque : pour un utilisateur dont le compte a été créé en 2014 ou avant, il est constitué par le mot de passe initial indiqué dans la dernière lettre reçue de l'IDRIS.
Ce mot de passe initial doit être changé dans les 20 jours qui suivent la réception du courriel contenant le mot de passe généré aléatoirement (c.f. ci-dessous le paragraphe intitulé Utilisation du mot de passe initial).
Passé ce délai de 20 jours, le mot de passe initial est invalidé et un courriel vous est envoyé pour vous en informer. Il suffit de contacter par courriel
pour demander la réinitialisation de la partie aléatoire du mot de passe initial, qui vous est alors renvoyée par courriel.
Le mot de passe initial est (ré)généré dans les cas suivants :
- Réouverture de compte : un mot de passe initial est attribué à la création de chaque compte, mais aussi lors de la réouverture d'un compte fermé.
- Oubli de votre mot de passe :
- Si vous perdez votre mot de passe courant, contactez par courriel pour demander la réinitialisation de la partie aléatoire du mot de passe initial, qui vous est alors renvoyée par courriel ; vous aurez aussi besoin de votre mot de passe initial utilisateur.
- Si vous avez aussi perdu votre mot de passe initial utilisateur, celui que vous avez vous-même fourni dans le FGC (ou reçu dans la lettre de l'IDRIS avant 2014), vous devez remplir dans le FGC le cadre de changement de mot de passe initial utilisateur, l'imprimer et le signer, puis le transmettre par courriel à ou l'envoyer par courrier postal. Vous recevrez alors un courriel dans lequel un nouveau mot de passe aléatoire vous sera communiqué.
Utilisation du mot de passe initial lors de la première connexion
Voici un exemple de première connexion avec entrée du mot de passe initial pour le compte login_idris sur la machine de l'IDRIS.
Attention : à la première connexion, le mot de passe initial est demandé deux fois. Une première fois pour établir la connexion sur la machine et une seconde fois par la procédure de changement du mot de passe qui est alors automatiquement exécutée.
Recommandation : comme vous êtes obligé de changer le mot de passe initial à votre première connexion, préparez soigneusement un autre mot de passe avant de commencer la procédure (c.f. Règles de constitution ci-dessous).
$ ssh login_idris@machine_idris.idris.fr login_idris@machine_idris password: ## Première demande du MOT DE PASSE INITIAL ## Last login: Fri Nov 28 10:20:22 2014 from machine_idris.idris.fr WARNING: Your password has expired. You must change your password now and login again! Changing password for user login_idris. Enter login( ) password: ## Seconde demande du MOT DE PASSE INITIAL ## New password: ## Entrez le nouveau mot de passe choisi ## Retype new password: ## Confirmez le nouveau mot de passe choisi ## password information changed for login_idris passwd: all authentication tokens updated successfully. Connection to machine_idris closed.
Remarque : le fait d'être immédiatement déconnecté après que le nouveau mot de passe choisi ait été accepté (all authentication tokens updated successfully) est normal.
Vous pouvez maintenant vous reconnecter avec le nouveau mot de passe courant que vous venez d'enregistrer.
Le mot de passe courant
Une fois choisi correctement, sa durée de validité est d'un an (365 jours).
Comment changer votre mot de passe courant ?
Vous pouvez changer votre mot de passe en utilisant la commande UNIX passwd sur la frontale de la machine. Le changement est immédiat et effectif sur tous les nœuds de la machine. La validité de ce nouveau mot de passe courant est aussi d'un an (365 jours).
Règles de constitution des mots de passe courants :
- Ils doivent comporter un minimum de 12 caractères.
- Ces caractères doivent appartenir à au moins 3 familles de caractères parmi les 4 suivantes :
- majuscules,
- minuscules,
- chiffres,
- caractères spéciaux.
- Un mot de passe ne doit pas non plus contenir le même caractère plus de deux fois consécutives.
- Il ne doit pas être composé de mots issus de dictionnaires, ni de combinaisons triviales (1234, azerty, …).
Remarques :
- Un nouveau mot de passe n'est pas modifiable pendant 5 jours. Il est toujours possible, en cas de nécessité, de contacter l'Assistance pour demander une remise à zéro au mot de passe initial.
- Un historique des 6 derniers mots de passe est conservé, afin de rejeter un mot de passe utilisé récemment.
Oubli ou expiration du mot de passe courant
Si vous avez oublié votre mot de passe courant ou si, malgré les courriels d'avertissement, vous n'avez pas changé votre mot de passe courant avant sa date d'expiration (un an après le dernier changement) alors votre mot de passe sera invalidé.
Vous devez alors contactez par courriel pour demander la réinitialisation de la partie aléatoire du mot de passe initial, qui vous est alors renvoyée par courriel.
Remarque : vous aurez aussi besoin de la partie utilisateur de votre mot de passe initial pour pouvoir vous connecter sur la machine après cette réinitialisation. En effet, vous aurez à suivre la procédure correspondant à une utilisation du mot de passe initial lors de la première connexion.
Compte bloqué suite à 15 connexions infructueuses :
Si votre compte est bloqué suite à 15 tentatives infructueuses, vous devez contacter l'Assistance de l'IDRIS.
Rappel sur la sécurité de votre compte
Ne communiquez jamais votre mot de passe en clair dans un message électronique, même ceux adressés à l'IDRIS (Assistance, Gestion des Utilisateurs, etc.) et ce, quel qu'en soit le motif : nous serions alors obligés de générer immédiatement un nouveau mot de passe initial, afin d'inhiber le mot de passe courant que vous auriez ainsi publié et de nous assurer que vous en définissiez un nouveau dès la connexion suivante.
Chaque compte est strictement personnel. L'accès à un compte par une personne non autorisée entraîne dès sa découverte des mesures de protection immédiates allant jusqu'au blocage du compte.
En tant que titulaire d'un compte, vous êtes tenus de prendre quelques précautions élémentaires de bon sens :
- prévenir immédiatement l'IDRIS de toute tentative de violation de son compte,
- respecter les recommandations sur l'utilisation des clés ssh,
- protéger ses fichiers en limitant les droits d'accès UNIX,
- ne pas utiliser de mot de passe trop simple,
- protéger son poste de travail personnel.
6. Gestion de son compte et de son environnement
Comment modifier mes données personnelles ?
La modification de vos données personnelles se fait via l'interface Web Extranet.
- Pour ceux qui n'ont pas de mot de passe Extranet ou qui l'ont perdu, les modalités d'accès sont décrites sur cette page.
- Pour ceux qui ont leur mot de passe, cliquez sur Extranet, connectez vous avec vos identifiants puis ⇒ Votre compte ⇒ Vos données ⇒ Coordonnées.
Les seules données modifiables en ligne sont :
- le courriel ;
- le téléphone ;
- le fax.
La modification de vos coordonnées postales se fait en nous envoyant le formulaire de gestion de compte FGC.
Quels espaces disques sont disponibles sur Jean Zay ?
Pour chaque projet, 5 espaces disques distincts sont disponibles sur Jean Zay : HOME, WORK, SCRATCH/JOBSCRATCH, STORE et DSDIR.
Vous trouverez les explications concernant ces espaces sur notre site web, à la page Espaces disques.
Attention : HOME, WORK et STORE sont soumis à des quotas !
Si votre login est rattaché à plusieurs projets, la commande IDRIS idrenv vous affichera toutes les variables d'environnement référençant tous les espaces disques de tous vos projets. Ces variables vous permettent d'accéder aux données de vos divers projets à partir de n'importe lequel d'entre eux.
Choisissez votre espace de stockage selon vos besoins (données permanentes, semi-temporaires, temporaires, gros ou petits fichiers, etc …).
Comment demander une extension d'espace disque ou d'inodes ?
Si votre utilisation de l'espace disque est conforme à son usage et si vous ne pouvez pas supprimer ou déplacer des données contenues dans cet espace, alors votre chef de projet peut effectuer une demande argumentée d'augmentation de quota (espace et/ou inodes) sur l'extranet de l'IDRIS.
Comment connaitre le nombre d'heures de calcul consommées par projet ?
Il vous suffit d'utiliser la commande IDRIS idracct pour connaitre les heures consommées par chaque collaborateur du projet ainsi que le pourcentage de l'allocation d'heures et le total consommés.
Notez que les informations retournées par cette commande ne sont mises à jour qu'une fois par jour (comme indiqué en première ligne de la sortie de la commande).
Si vous avez plusieurs projets à l'IDRIS, cette commande vous donnera les consommations CPU et/ou GPU de tous les projets auxquels votre login est rattaché.
Que faire lorsque je n'ai bientôt plus d'heures de calcul ?
Il est possible d'effectuer des demandes d'heures supplémentaires :
- soit une demande dite “au fil de l'eau” qui peut se faire tant que votre projet AD ou IA dispose d'une allocation d'heures en cours.
- soit une demande à mi-parcours de votre allocation d'Accès Régulier dans le cas d'un AR pour obtenir un complément d'heures pour une période de six mois.
Ces demandes, qui doivent être justifiées, sont à effectuer via le portail eDARI comme indiqué dans notre page web traitant des demandes d'allocations d'heures.
Comment puis-je savoir si la machine est indisponible ?
La machine peut être indisponible en raison d'une maintenance préprogrammée ou d'un problème technique survenu subitement. Dans les deux cas, l'information est disponible sur la page d'accueil du site Web de l'IDRIS via le menu déroulant intitulé “Espace utilisateurs” puis la rubrique "Disponibilité des machines".
Les utilisateurs de l'IDRIS peuvent également s'inscrire à la liste de diffusion “info-machines” via l'Extranet. .
Comment récupérer des fichiers que j'ai malencontreusement détruits ?
Suite à la migration vers les nouveaux espaces de stockage Lustre, l'espace disque WORK n'est plus sauvegardé. Nous vous recommandons de conserver une copie de vos données importantes sous forme d'archives stockées sur votre STORE.
Pour des raisons de trop grande taille, ni le SCRATCH (espace semi-temporaire), ni le STORE (espace de stockage) ne sont sauvegardés.
Puis-je demander à l'IDRIS le transfert des fichiers d'un compte sur un autre compte ?
L'IDRIS considère que les données sont liées à un projet. Par conséquent, pour que le transfert soit possible, il faut que :
- les 2 comptes (le propriétaire et le destinataire) soient tous les deux rattachés au même projet ;
- que ce soit le chef du projet qui fasse la demande, par fax signé ou par courriel à l'assistance () en précisant bien :
- la machine concernée ;
- le compte source et le compte destinataire ;
- la liste des fichiers et/ou répertoires à transférer.
Puis-je récupérer des fichiers sur un support externe ?
Il n'est plus possible de demander le transfert de ses fichiers sur un support externe.
7. Présentation des espaces disques
Jean Zay : les espaces disques
Pour chaque projet, quatre espaces disques distincts sont accessibles : HOME, WORK et SCRATCH/JOBSCRATCH, enfin le STORE.
Chaque espace a des caractéristiques spécifiques adaptées à son utilisation, qui sont décrites ci-dessous. Les chemins d'accès à ces espaces sont stockés dans cinq variables d'environnement du shell : $HOME, $WORK, $SCRATCH et $JOBSCRATCH, enfin $STORE.
Vous pouvez connaître l'occupation des différents espaces disques avec les commandes IDRIS “idr_quota_user/idr_quota_project” ou avec la commande Unix du (disk usage). Le retour des commandes idr_quota_user et idr_quota_project est immédiat mais n'est pas une information en temps réel (les données sont actualisées une fois par jour). La commande du retourne une information en temps réel mais son exécution peut prendre beaucoup de temps selon la taille du répertoire concerné.
Pour la gestion spécifique de bases de données sur Jean Zay, une page dédiée a été rédigée en complément de celle-ci : Gestion de bases de données.
Le HOME
$HOME : c'est le répertoire d'accueil lors d'une connexion interactive. Cet espace est destiné aux fichiers de petite taille, très souvent utilisés, comme les fichiers d'environnement du shell, les utilitaires, éventuellement les sources et les bibliothèques quand leur taille est raisonnable. Cet espace a une taille limitée (en espace comme en nombre de fichiers).
Voici ses caractéristiques :
- le HOME est un espace permanent ;
- il est prévu pour accueillir des fichiers de petite taille ;
- dans le cas d'un login multi-projets, le HOME est unique ;
- il est soumis à des quotas par utilisateur volontairement assez faibles (3 Gio par défaut) ;
- il est accessible en interactif ou dans un travail batch via la variable
$HOME:$ cd $HOME
- c'est le répertoire d'accueil lors d'une connexion interactive.
Remarque : l'espace HOME est aussi référencé via la variable d'environnement CCFRHOME pour respecter une nomenclature commune avec les autres centres de calcul nationaux (CINES, TGCC) :
$ cd $CCFRHOME
Le WORK
$WORK : c'est un espace de travail et de stockage permanent utilisable en batch. On y stocke généralement les fichiers de taille importante qui servent lors des exécutions en batch : les fichiers sources volumineux et les bibliothèques, les fichiers de données, les exécutables, les fichiers de résultats, les scripts de soumission.
Voici ses caractéristiques :
- le WORK est un espace permanent ;
- il est prévu pour accueillir des fichiers de taille importante : la taille maximum est de 10 Tio par fichier ;
- dans le cas d'un login multi-projet, un WORK par projet est créé ;
- il est soumis à des quotas par projet ;
- il est accessible en interactif ou dans un travail batch ;
- il est composé de 2 parties :
- une partie propre à chaque utilisateur ; on y accède par la commande :
$ cd $WORK
- une partie commune au projet auquel l'utilisateur appartient, dans lequel on peut mettre des fichiers destinés à être partagés; on y accède par la commande :
$ cd $ALL_CCFRWORK
- le WORK est un espace disque dont la bande passante est d'environ 100 Go/s en écriture et en lecture. Celle-ci peut être ponctuellement saturée en cas d'utilisation exceptionnellement intensive.
Remarque : l'espace WORK est aussi référencé via la variable d'environnement CCFRWORK pour respecter une nomenclature commune avec les autres centres de calcul nationaux (CINES, TGCC) :
$ cd $CCFRWORK
Recommandations d'utilisation :
- les travaux batch peuvent s'exécuter dans le WORK ; cependant, plusieurs de vos travaux pouvant s'exécuter en même temps, il vous faut gérer l'unicité de vos répertoires d'exécution ou de vos noms de fichiers.
- De plus, il est soumis à des quotas (par projet) qui peuvent stopper brutalement votre exécution s'ils sont atteints. Ainsi, dans le WORK, il faut tenir compte non seulement de votre propre activité, mais aussi de celle de vos collègues de projet. Pour ces raisons, on pourra alors être amené à lui préférer le SCRATCH ou le JOBSCRATCH pour l'exécution de ses travaux batch.
Le SCRATCH/JOBSCRATCH
$SCRATCH : c'est un espace de travail et de stockage semi-temporaire utilisable en batch, la durée de vie des fichiers y est limitée à 30 jours. On y stocke généralement les fichiers de taille importante qui servent lors des exécutions en batch : les fichiers de données, les fichiers de résultats ou de reprise de calcul (restarts). Une fois le post-traitement effectué pour réduire le volume de données, il faut penser à recopier les fichiers significatifs dans le WORK pour ne pas les perdre après 30 jours, ou dans le STORE pour un archivage à long terme.
Voici ses caractéristiques :
- le SCRATCH est un espace semi-permanent : la durée de vie des fichiers est de 30 jours ;
- il n'est pas sauvegardé ;
- il est prévu pour accueillir des fichiers de taille importante : la taille maximum est de 10 Tio par fichier ;
- il est soumis à des quotas de sécurité très larges :
- des quotas disque par projet de l'ordre d'1/10ième de l'espace disque total
- et des quotas inode par projet de l'ordre de 150 millions de fichiers et répertoires;
- il est accessible en interactif ou dans un travail batch ;
- il est composé de 2 parties :
- une partie propre à chaque utilisateur ; on y accède par la commande :
$ cd $SCRATCH
- une partie commune au projet auquel l'utilisateur appartient, dans lequel on peut mettre des fichiers destinés à être partagés; on y accède par la commande :
$ cd $ALL_CCFRSCRATCH
- dans le cas d'un login multi-projet, un SCRATCH par projet est créé ;
- le SCRATCH est un espace disque dont la bande passante est d'environ 500 Go/s en écriture et en lecture.
Remarque : l'espace SCRATCH est aussi référencé via la variable d'environnement CCFRSCRATCH pour respecter une nomenclature commune avec les autres centres de calcul nationaux (CINES, TGCC) :
$ cd $CCFRSCRATCH
$JOBSCRATCH : c'est le répertoire temporaire d'exécution propre à un unique travail batch.
Voici ses caractéristiques :
- le JOBSCRATCH est un répertoire temporaire : la durée de vie des fichiers est celle du travail batch ;
- il n'est pas sauvegardé ;
- il est prévu pour accueillir des fichiers de taille importante : la taille maximum est de 10 Tio par fichier ;
- il est soumis à des quotas de sécurité très larges :
- des quotas disque par projet de l'ordre d'1/10ième de l'espace disque total
- et des quotas inode par projet de l'ordre de 150 millions de fichiers et répertoires;
- il est créé automatiquement lorsqu'un travail batch démarre : il est donc unique à chaque travail ;
- il est détruit automatiquement à la fin de ce travail : il est donc indispensable de recopier explicitement les fichiers importants sur un autre espace disque (le WORK ou le SCRATCH) avant la fin du travail ;
- le JOBSCRATCH est un espace disque dont la bande passante est d'environ 500 Go/s en écriture et en lecture.
- Pendant toute la durée d'exécution d'un travail batch, le JOBSCRATCH correspondant est accessible depuis la frontale Jean Zay via son numéro de job JOBID (voir la sortie de la commande squeue), votre login (variable d'environnement LOGNAME) et la commande suivante :
$ cd /lustre/fsn1/jobscratch_hpe/$LOGNAME_JOBID
Recommandations d'utilisation :
- On peut voir le JOBSCRATCH comme l'ancien TMPDIR.
- Le SCRATCH peut être vu comme un WORK semi-temporaire, mais avec les performances d’entrées/sortie maximales offertes à l'IDRIS, au prix d'une durée de vie des fichiers de 30 jours.
- Les caractéristiques semi-temporaires du SCRATCH permettent d'y stocker de gros volumes de données entre deux ou plusieurs jobs qui s’enchainent sur une durée limitée à quelques semaines : cet espace n'est pas “purgé” après chaque job.
Le STORE
$STORE : c'est l'espace d'archivage de l'IDRIS destiné au stockage à long terme de données. On y stocke généralement les fichiers de taille très importante, fruits du tar d'une arborescence de fichiers résultats de calcul, après post-traitement. C'est un espace qui n'a pas pour vocation d’être accédé ou modifié quotidiennement, mais de préserver dans le temps de très gros volumes de données avec une consultation épisodique.
Changement important : Depuis le 22 juillet 2024, le STORE est uniquement accessible depuis les frontales et les partitions prepost, archive, compil et visu. Les travaux s'exécutant sur les nœuds de calcul n'auront plus directement accès à cet espace mais vous pouvez utiliser des travaux chaînés pour automatiser la gestion des données depuis/vers le STORE (voir nos exemples de travaux chaînés utilisant le STORE).
Voici ses caractéristiques :
- le STORE est un espace permanent ;
- il n'est pas accessible depuis les nœuds de calcul mais uniquement depuis les frontales et les partitions prepost, archive, compil et visu (vous pouvez utiliser des travaux chaînés pour automatiser la gestion des données depuis/vers le STORE, voir nos exemples de travaux chaînés utilisant le STORE) ;
- il est prévu pour accueillir des fichiers de taille très importante : la taille maximum est de 10 Tio par fichier et la taille minimum conseillée de 250 Mio (ratio taille disque / nombre d'inodes);
- dans le cas d'un login multi-projet, un STORE par projet est créé ;
- il est soumis à des quotas par projet avec un faible nombre d'inodes, mais un très grand espace ;
- il est composé de 2 parties :
- une partie propre à chaque utilisateur ; on y accède par la commande :
$ cd $STORE
- une partie commune au projet auquel l'utilisateur appartient, dans lequel on peut mettre des fichiers destinés à être partagés; on y accède par la commande :
$ cd $ALL_CCFRSTORE
Remarque : l'espace STORE est aussi référencé via la variable d'environnement CCFRSTORE pour respecter une nomenclature commune avec les autres centres de calcul nationaux (CINES, TGCC) :
$ cd $CCFRSTORE
Recommandations d'utilisation :
- Le STORE n'a pas de principe de limitation de la durée de vie des fichiers.
- Comme il s'agit d'un espace d'archive, il n'est pas conçu pour des accès fréquents.
Le DSDIR
$DSDIR : l'espace disque contenant les bases de données publiques volumineuses (en taille ou en nombre de fichiers) nécessaires à l'utilisation d'outils de l'Intelligence Artificielle. Ces bases de données sont visibles par l'ensemble des utilisateurs de Jean Zay.
Si vous exploitez de telles bases de données et que celles-ci ne se trouvent pas dans l'espace $DSDIR, l'IDRIS les téléchargera et les installera dans cet espace disque à votre demande.
Vous trouverez la liste des bases de données disponibles actuellement sur la page : Jean Zay : jeux de données et modèles disponibles dans l'espace de stockage $DSDIR.
Si votre base de données est personnelle ou sous licence trop restrictive, il vous faudra prendre en charge vous-même sa gestion sur les espaces disques de votre projet, comme décrit dans la page Gestion de bases de données.
Tableau récapitulatif des principaux espaces disques
| Espace | Capacité par défaut | Spécificité | Usage |
|---|---|---|---|
$HOME |
3 Go et 150 kinodes par utilisateur |
- Accueil de connexion | - Stockage de fichiers de configuration et de petits fichiers |
$WORK |
5 To (*) et 500 kinodes par projet |
- Stockage sur disques rotatifs (350 Go/s en lecture et 300 Go/s en écriture) |
- Stockage des sources et des données d'entrée/sortie - Exécution en batch ou interactif |
$SCRATCH |
Quotas de sécurité très larges 4,6 Po partagés par tous les utilisateurs | - Espace non sauvegardé - Stockage SSD (1,5 To/s en lecture et 1,1 To/s en écriture) - Durée de vie des fichiers inutilisés : 30 jours (inutilisés = non lus, non modifiés) |
- Stockage des données d'entrée/sortie volumineuses - Exécution en batch ou interactif - Performances optimales pour les opérations de lecture/écriture |
$STORE |
50 To (*) et 100 kinodes (*) par projet |
- Cache disque et bandes magnétiques - Accès longs si fichier uniquement sur bande. |
- Stockage d'archives sur du long terme (durée de vie du projet) - Pas accessible depuis les nœuds de calcul |
Les sauvegardes
ATTENTION : Suite à la migration vers les nouveaux espaces de stockage Lustre, l'espace disque WORK n'est plus sauvegardé. Nous vous recommandons de conserver une copie de vos données importantes sous forme d'archives stockées sur votre STORE.
Jean Zay : quotas disques et commandes de visualisation des taux d'occupation
Principe
Les quotas garantissent un accès équitable aux ressources disques. Ils évitent qu'un groupe d'utilisateurs ne consomme tout l'espace et n'empêche les autres groupes de travailler. À l'IDRIS, les quotas limitent à la fois la quantité d'espace disque et le nombre de fichiers (inodes). Ces limites s'appliquent par utilisateur pour votre HOME (un HOME par utilisateur même si votre login est rattaché à plusieurs projets) et par projet pour le WORK et le STORE (autant de WORK et de STORE que de projets pour un même utilisateur).
Vous pouvez consulter l'occupation de vos espaces disques en utilisant l'une des deux commandes présentées dans cette page :
idr_quota_userpour une vue de votre utilisation personnelle en tant qu'utilisateur ;idr_quota_projectpour une vue d'ensemble de votre projet et de la consommation de chacun de ses membres.
La commande idrquota n'est plus disponible. Il s'agissait de la première commande de visualisation de quotas qui avait été déployée sur Jean Zay. Les commandes idr_quota_user et idr_quota_project en sont une évolution.
Dépassement des quotas
Lorsqu'un utilisateur ou un projet est en dépassement de quota, aucun email d'avertissement n'est envoyé. Vous êtes néanmoins informé par des messages d'erreur du type Disk quota exceeded lorsque vous manipulez des fichiers dans l'espace disque concerné.
Lorsque l'un des quotas est atteint, vous ne pouvez plus créer de fichiers dans l'espace disque concerné. Cela peut donc perturber les travaux en cours d'exécution s'ils ont été lancés depuis cet espace.
Attention, éditer un fichier en étant en limite de quota disque peut ramener sa taille a zéro, donc en effacer le contenu.
Lorsque vous êtes bloqués ou en passe de l’être :
- Essayez de faire du ménage dans l'espace disque concerné en supprimant les fichiers devenus obsolètes.
- Archivez les répertoires auxquels vous n'accédez plus ou rarement.
- Déplacez vos fichiers dans un autre espace en fonction de leurs usages (voir la page sur les espaces disques).
- Le chef de projet ou son suppléant peuvent demander une augmentation des quotas de l'espace STORE via l'interface Extranet.
Remarques:
- Il faut penser à vérifier les espaces disques communs
$ALL_CCFRWORKet$ALL_CCFRSTORE. - Une cause récurrente de dépassement de quota est l'utilisation d'environnements conda personnels. Veuillez vous référer à la page Environnement Python personnel pour connaître les bonnes pratiques sur Jean Zay.
La commande idr_quota_user
Par défaut, la commande idr_quota_user renvoie votre occupation personnelle en tant qu'utilisateur pour l'ensemble des espaces disques de votre projet actif. Par exemple, si votre projet actif est abc, vous verrez une sortie similaire à celle-ci :
$ idr_quota_user HOME INODE: |██-------------------------------| U: 9419/150000 6.28% STORAGE: |████████████████████████████████-| U: 2.98 GiB/3.00 GiB 99.31% ABC STORE INODE: |---------------------------------| U: 1/100000 0.00% G: 12/100000 0.01% STORAGE: |---------------------------------| U: 4.00 KiB/50.00 TiB 0.00% G: 48.00 KiB/50.00 TiB 0.00% ABC WORK INODE: |███▒▒▒---------------------------| U: 50000/500000 10.00% G: 100000/500000 20.00% STORAGE: |██████████▒▒▒▒▒▒▒▒▒▒-------------| U: 1.25 TiB/5.00 TiB 25.00% G: 2.5 TiB/5.00 TiB 50.00% The quotas are refreshed daily. All the information is not in real time and may not reflect your real storage occupation.
Sur cet exemple de sortie, votre occupation personnelle est représentée par la barre noire et quantifiée sur la droite après la lettre U (pour User).
Votre occupation personnelle est aussi comparée à l'occupation globale du projet qui est représentée par la barre grise (sur cet exemple de sortie). et quantifiée après la lettre G (pour Groupe).
Notez que les couleurs peuvent être différentes suivant les paramètres et/ou le type de votre terminal.
Vous pouvez affiner l'information retournée par la commande idr_quota_user en ajoutant un ou plusieurs des arguments suivants :
--project defpour afficher l'occupation d'un projet différent de votre projet actif (icidef) ;--all-projectspour afficher l'occupation de l'ensemble des projets auxquels vous êtes rattaché ;--space home workpour afficher l'occupation d'un (ou plusieurs) espace(s) disque(s) en particulier (ici le HOME et le WORK).
L'aide complète de la commande idr_quota_user est accessible en lançant :
$ idr_quota_user -h
La commande idr_quota_project
Par défaut, la commande idr_quota_project renvoie l'occupation disque de chaque membre de votre projet actif pour l'ensemble des espaces disques associés au projet. Par exemple, si votre projet actif est abc, vous verrez une sortie similaire à celle-ci :
$ idr_quota_project PROJECT: abc SPACE: WORK PROJECT USED INODE: 34373/500000 6.87% PROJECT USED STORAGE: 1.42 GiB/5.00 TiB 0.03% ┌─────────────────┬─────────────────┬─────────────────┬─────────────────┬──────────────────────┐ │ LOGIN │ INODE ▽ │ INODE % │ STORAGE │ STORAGE % │ ├─────────────────┼─────────────────┼─────────────────┼─────────────────┼──────────────────────┤ │ abc001 │ 29852│ 5.97%│ 698.45 MiB│ 0.01%│ │ abc002 │ 4508│ 0.90%│ 747.03 MiB│ 0.01%│ │ abc003 │ 8│ 0.00%│ 6.19 MiB│ 0.00%│ │ abc004 │ 1│ 0.00%│ 0.00 B│ 0.00%│ │ abc005 │ 1│ 0.00%│ 0.00 B│ 0.00%│ └─────────────────┴─────────────────┴─────────────────┴─────────────────┴──────────────────────┘ PROJECT: abc SPACE: STORE PROJECT USED INODE: 13/100000 0.01% PROJECT USED STORAGE: 52.00 KiB/50.00 TiB 0.00% ┌─────────────────┬─────────────────┬─────────────────┬─────────────────┬──────────────────────┐ │ LOGIN │ INODE ▽ │ INODE % │ STORAGE │ STORAGE % │ ├─────────────────┼─────────────────┼─────────────────┼─────────────────┼──────────────────────┤ │ abc001 │ 2│ 0.00%│ 8.00 KiB│ 0.00%│ │ abc002 │ 2│ 0.00%│ 8.00 KiB│ 0.00%│ │ abc003 │ 2│ 0.00%│ 8.00 KiB│ 0.00%│ │ abc004 │ 2│ 0.00%│ 8.00 KiB│ 0.00%│ │ abc005 │ 1│ 0.00%│ 4.00 KiB│ 0.00%│ └─────────────────┴─────────────────┴─────────────────┴─────────────────┴──────────────────────┘ The quotas are refreshed daily. All the information is not in real time and may not reflect your real storage occupation.
Pour chaque espace disque, un résumé de l'occupation globale est affiché, suivi d'un tableau détaillé de l'occupation de chaque membre du projet.
Vous pouvez affiner l'information retournée par la commande idr_quota_project en ajoutant un ou plusieurs des arguments suivants :
--project defpour afficher l'occupation d'un projet différent de votre projet actif (icidef) ;--space workpour afficher l'occupation d'un (ou plusieurs) espace(s) disque(s) en particulier (ici le WORK) ;--order storagepour trier les informations dans l'ordre décroissant des valeurs d'une colonne donnée (ici la colonne STORAGE)
L'aide complète de la commande idr_quota_project est accessible en lançant :
$ idr_quota_project -h
Remarques générales
- Les projets auxquels vous êtes rattaché correspondent aux groupes UNIX listés par la commande
idrproj. - Les quotas ne sont pas relevés en temps réel et ne représentent peut-être pas l'état actuel d'occupation de vos espaces disques. Les commandes
idr_quota_useretidr_quota_projectsont actualisées tous les matins. - Pour connaître en temps réél la volumétrie en octet et inodes d'un répertoire donné
my_directory, vous pouvez exécuter les commandesdu -hd0 my_directoryetdu -hd0 --inodes my_directory, respectivement. Contrairement aux commandesidr_quota, le temps d'exécution des commandesdupeut être important, relativement à la taille du répertoire. - Pour le WORK et le STORE, les taux d'occupation affichés englobent à la fois l'occupation de l'espace personnel (
$WORKou$STORE) et l'occupation de l'espace commun ($ALL_CCFRWORKou$ALL_CCFRSTORE).
8. Commandes de transferts de fichiers
Les transferts de fichiers par la commande bbftp
Pour transférer des fichiers volumineux de l'IDRIS vers votre laboratoire, nous vous conseillons d'utiliser BBFTP qui est un logiciel optimisé pour le transfert de fichiers.
Toutes les informations sur l'utilisation de la commande bbftp se trouvent notre site Web.
Les transferts de fichiers via le réseau CCFR
Comment transférer des données entre les 3 centres nationaux (réseau CCFR) ?
Présentation
Le réseau des Centres de Calcul Français (CCFR) est un réseau dédié à très haut débit, qui interconnecte les trois centres de calcul nationaux CINES, IDRIS et TGCC. Ce réseau est mis à la disposition des utilisateurs pour faciliter les transferts de données entre les centres nationaux. Sont actuellement raccordées sur ce réseau les machines Joliot-Curie au TGCC, Jean Zay à l'IDRIS et Adastra au CINES.
L'utilisation de ce réseau implique que vous ayez des logins (différents pour chaque centre) dans au moins deux des trois centres et qu'ils soient autorisés à accéder au réseau CCFR dans les centres concernés.
Remarques :
- Pour votre login IDRIS, la demande d'accès au réseau CCFR peut se faire :
- lors de votre demande de création de compte à partir du portail eDARI,
- ou en envoyant un mail à à partir de votre adresse connue du centre avec comme titre « CCFR : login IDRIS / votre nom » pour demander l’accès CCFR et vous permettre de faire vos transferts vers le CINES et le TGCC. L’information sera transmise aux deux autres centres pour qu'ils puissent aussi faire ce qui est nécessaire pour que cela soit opérationnel.
- De plus, tous les nœuds de Jean Zay ne sont pas connectés à ce réseau. Pour l'utiliser depuis l'IDRIS, vous pouvez utiliser les frontales
jean-zay.idris.fretjean-zay-pp.idris.fr.
Pour plus d'information, n'hésitez pas à contacter l'assistance ().
Transfert de données via CCFR
Le transfert de données entre les machines des centres via le réseau CCFR constitue le service principal pour ce réseau. Une commande wrapper ccfr_cp accessible par l'intermédiaire d'un module est fournie pour simplifier les usages :
$ module load ccfr
Cette commande ccfr_cp récupère automatiquement les informations de connexion à la machine spécifiée (nom de domaine, port spécifique) et détecte les possibilités d’authentification. Par défaut, la commande optera pour une authentification basique, utilisant les modalités traditionnelles en vigueur sur la machine ciblée.
La commande ccfr_cp est basée sur l’outil rsync, configuré pour faire transiter les données avec le protocole SSH. La copie réalisée est récursive et préserve les liens symboliques, les droits d’accès ainsi que les dates de modification des fichiers.
Le détail de la commande, ainsi que la liste des machines accessibles sur le réseau CCFR sont disponibles en précisant l’option -h à la commande ccfr_cp.
Pour des transferts depuis jean-zay vers les machines du CINES et du TGCC, vous pouvez utiliser des commandes similaires à celles-ci :
$ module load ccfr $ ccfr_cp /path/to/datas/on/jean-zay login_cines@adastra:/path/to/directory/on/adastra: $ ccfr_cp /path/to/datas/on/jean-zay login_tgcc@irene:/path/to/directory/on/irene:
Pour effectuer des transferts depuis Adastra, la procédure est similaire excepté que vous devez utiliser la machine adastra-ccfr.cines.fr (accessible depuis adastra.cines.fr) comme indiqué sur la documentation du CINES.
Pour effectuer des transferts depuis Joliot-Curie, la procédure est aussi similaire et peut être effectuée directement depuis la frontale irene-fr.ccc.cea.fr. Après connexion à la machine, la commande machine.info vous donnera toutes les informations utiles.
Une commande ccfr_sync, variante de ccfr_cp, permet une synchronisation forte entre la source et la destination en ajoutant, par rapport à la commande ccfr_cp, la suppression des fichiers de la destination qui ne sont pas présents dans la source.
Remarque : Par défaut, ces commandes utiliseront une authentification basique par mot de passe en respectant les modalités en vigueur du centre distant (CINES ou TGCC). Vous serez donc certainement obligés de fournir un mot de passe à chaque fois. Pour éviter ceci, vous pouvez utiliser les certificats IDRIS de type transfert-only (validité 7 jours) dont le mode d'emploi est défini sur le site web de l'IDRIS. L'utilisation de tels certificats vous obligera à initier les transferts depuis la machine distante adastra-ccfr.cines.fr (accessible depuis adastra.cines.fr) pour le CINES et irene-fr.ccc.cea.fr pour le TGCC après avoir copié le certificat transfert-only sur la machine distante et à construire vous-même les commandes rsync de transfert (donc ne pas utiliser les wrappers ccfr_cp et ccfr_sync). Vous pouvez alors vous inspirer des exemples suivants pour faire vos transferts :
# Simple copie de jean-zay vers machine distante (exécuté sur la machine distante) # utilisant le certificat transfert-only contenu dans ~/.ssh/id_ecc_rsync sur la machine distante $ rsync --human-readable --recursive --links --perms --times --omit-dir-times -v \ -e 'ssh -i ~/.ssh/id_ecc_rsync' \ login_idris@jean-zay-ccfr.idris.fr:/path/on/jean-zay /path/on/adastra/or/irene # Synchronisation forte (option --delete) de jean-zay vers machine distante (exécuté sur la machine distante) # utilisant le certificat transfert-only contenu dans ~/.ssh/id_ecc_rsync sur la machine distante $ rsync --human-readable --recursive --links --perms --times --omit-dir-times -v --delete \ -e 'ssh -i ~/.ssh/id_ecc_rsync' \ login_idris@jean-zay-ccfr.idris.fr:/path/on/jean-zay /path/on/adastra/or/irene
Attention : Sur la machine adastra-ccfr.cines.fr, le certificat id_ecc_rsync doit être visible depuis votre repertoire /home/login_cines/.ssh pour que la commande ssh puisse l'utiliser (pas de variable d'environnement définie pour cet espace disque). Il faut donc prendre soin de dé-archiver le certificat dans ce repertoire avec une commande du style :
login_cines@adastra-ccfr.cines.fr:~$ unzip ~/transfert_certif.zip -d /home/login_cines/.ssh Archive: /lus/home/.../transfert_certif.zip inflating: /home/login_cines/.ssh/id_ecc_rsync inflating: /home/login_cines/.ssh/id_ecc_rsync.pub
Transfert de données via parallel-sftp
Pour accélérer les transferts de fichiers entre trois centres nationaux, vous pouvez également utiliser la commande parallel-sftp. Il s'agit d'un outil développé par le CEA qui permet d'utiliser la commande sftp standard en parallèle pour réaliser des transferts de fichiers parallèles.
Vous trouverez plus d'informations sur le site du CEA.
parallel-sftp est installé sur les nœuds de connexion du cluster IDRIS jean-zay. Son utilisation est identique à sftp, à l'exception de l'option -n qui permet de choisir le nombre de connexions ssh utilisées pour les transferts parallèles.
Par exemple, pour réaliser un transfert parallèle avec 5 connexions ssh :
$ parallel-sftp -n 5 <remote_login>@<remote_host>
Ainsi, si un transfert sftp est limité à 1 Gbps par exemple, ce transfert utilisera au maximum 5 Gbps.
Attention : pour vos transferts entre les centres nationaux, vous devez utiliser les nœuds connectés au réseau CCFR. Pour l'IDRIS, cela signifie que le paramètre <remote_host> ci-dessus doit être adastra-ccfr.cines.fr ou irene-fr-ccfr.ccc.cea.fr. Mais vous pouvez directement utiliser jean-zay.idris.fr pour exécuter la commande parallel-sftp.
Pour une utilisation à l'IDRIS, vous pouvez suivre l'exemple suivant écrit pour adastra (pour irene il suffit d'utiliser irene-fr-ccfr.ccc.cea.fr):
# 1/ Se connecter à jean-zay : $ ssh jean-zay_login@jean-zay.idris.fr # 2/ Pour initier une connexion permettant de faire des transferts avec la machine adastra # utilisant potentiellement 5 threads dans ce cas $ parallel-sftp -n 5 adastra_login@adastra-ccfr.cines.fr sftp> # Vous êtes alors dans un environnement sftp dans lequel pour pouvez exécuter # des commandes put et get pour faire les transferts (voir man sftp). # ATTENTION : les variables d'environnement telles que WORK, SCRATCH, STORE, ... # ne sont pas définies et ne peuvent donc pas être utilisées pour # spécifier les chemins d'accès aux données à transférer. # Vous devez indiquer les chemins complets ! # 3/ Faire les transferts # Pour transférer un répertoire de jean-zay sur adastra, vous pouvez faire : sftp> put -r /path/to/jean-zay/src/directory /path/to/adastra/dest/directory # Pour transférer un répertoire de adastra sur jean-zay, vous pouvez faire : sftp> get -r /path/to/jean-zay/dest/directory /path/to/adastra/src/directory
9. La commande module
Pour plus d’informations, consultez notre page web concernant l'utilisation de la commande module sur Jean Zay.
10. Compilation
Jean Zay : Appel du système de compilation Fortran et C/C++ (Intel)
$ module avail intel-compilers ----------------------- /gpfslocalsup/pub/module-rh/modulefiles -------------------------- intel-compilers/16.0.4 intel-compilers/18.0.5 intel-compilers/19.0.2 intel-compilers/19.0.4 $ module load intel-compilers/19.0.4 $ module list Currently Loaded Modulefiles: 1) intel-compilers/19.0.4
$ ifort prog.f90 -o prog $ icc prog.c -o prog $ icpc prog.C -o prog
Jean Zay : compilation d'un code parallèle MPI en Fortran, C/C++
- Intel MPI :
$ module avail intel-mpi -------------------------------------------------------------------------- /gpfslocalsup/pub/module-rh/modulefiles -------------------------------------------------------------------------- intel-mpi/5.1.3(16.0.4) intel-mpi/2018.5(18.0.5) intel-mpi/2019.4(19.0.4) intel-mpi/2019.6 intel-mpi/2019.8 intel-mpi/2018.1(18.0.1) intel-mpi/2019.2(19.0.2) intel-mpi/2019.5(19.0.5) intel-mpi/2019.7 intel-mpi/2019.9 $ module load intel-compilers/19.0.4 intel-mpi/19.0.4
- Open MPI (sans MPI CUDA-aware, il faudra sélectionner un module sans l'extension
-cuda) :
$ module avail openmpi -------------------------------------------------------- /gpfslocalsup/pub/modules-idris-env4/modulefiles/linux-rhel8-skylake_avx512 -------------------------------------------------------- openmpi/3.1.4 openmpi/3.1.5 openmpi/3.1.6-cuda openmpi/4.0.2 openmpi/4.0.4 openmpi/4.0.5 openmpi/4.1.0 openmpi/4.1.1 openmpi/3.1.4-cuda openmpi/3.1.6 openmpi/4.0.1-cuda openmpi/4.0.2-cuda openmpi/4.0.4-cuda openmpi/4.0.5-cuda openmpi/4.1.0-cuda openmpi/4.1.1-cuda $ module load pgi/20.4 openmpi/4.0.4
- Intel MPI :
$ mpiifort source.f90 $ mpiicc source.c $ mpiicpc source.C
- Open MPI :
$ mpifort source.f90 $ mpicc source.c $ mpic++ source.C
Jean Zay : compilation d'un code parallèle OpenMP en Fortran, C/C++
$ ifort -qopenmp source.f90 $ icc -qopenmp source.c $ icpc -qopenmp source.C
$ ifort -c -qopenmp source1.f $ ifort -c source2.f $ icc -c source3.c $ ifort -qopenmp source1.o source2.o source3.o
Jean Zay : Appel du système de compilation NVIDIA/PGI pour C/C++ et Fortran
$ module avail pgi nvidia-compilers --------------------- /gpfslocalsup/pub/module-rh/modulefiles --------------------- pgi/19.10 pgi/20.1 pgi/20.4 --------------------- /gpfslocalsup/pub/module-rh/modulefiles --------------------- nvidia-compilers/20.7 nvidia-compilers/21.7 nvidia-compilers/23.9 nvidia-compilers/20.9 nvidia-compilers/21.9 nvidia-compilers/23.11 nvidia-compilers/20.11 nvidia-compilers/22.5 nvidia-compilers/24.3 nvidia-compilers/21.3 nvidia-compilers/22.9 nvidia-compilers/21.5 nvidia-compilers/23.1 $ module load nvidia-compilers/24.3 $ module list Currently Loaded Modulefiles: 1) nvidia-compilers/24.3
$ nvc prog.c -o prog $ nvc++ prog.cpp -o prog $ nvfortran prog.f90 -o prog
Jean Zay : Compilation d'un code OpenACC
Les options de compilation pour la prise en charge d'OpenACC par le compilateur PGI sont les suivantes :
-acc: cette option active le support d'OpenACC. Il est possible de lui donner les sous-options suivantes :[no]autopar: Active la parallélisation automatique pour la directive ACC PARALLEL. Le défaut est de l'activer.[no]routineseq: Compile toutes les routines pour l'accélérateur. Le défaut est de ne pas compiler chaque routine comme séquentielle.strict: Affiche des messages d'avertissement en cas d'utilisation de directives non OpenACC pour l'accélérateur.verystrict: Arrête la compilation en cas d'utilisation de directives non OpenACC pour l'accélérateur.sync: Ignore les clauses async.[no]wait: Attend la fin de chaque noyau de calcul sur l'accélérateur. Le lancement de noyaux est bloqué par défaut, sauf si async est utilisé.- Exemple :
$ pgfortran -acc=noautopar,sync -o prog_ACC prog_ACC.f90
-ta: Cette option active le déport de calcul sur l'accélérateur. Elle implique l'option-acc.- Elle est utile pour choisir l'architecture cible pour laquelle le code va être compilé.
- Pour utiliser les GPU V100 de Jean Zay, il faut utiliser la sous-option
teslade-taet la compute capabilitycc70. Par exemple :$ pgfortran -ta=tesla:cc70 -o prog_gpu prog_gpu.f90
- Quelques exemples utiles de sous-options de
tesla:managed: Permet de créer une vue partagée de la mémoire des GPU et des CPU.pinned: Active l'épinglage de la mémoire sur le CPU. Cela peut améliorer les performances des transferts de données.autocompare: Active la comparaison des résultats sur CPU et GPU.
Jean Zay : MPI CUDA-aware et GPUDirect
Pour une performance optimale, des bibliothèques OpenMPI CUDA-aware supportant le GPUDirect sont disponibles sur Jean Zay.
$ module avail openmpi/*-cuda ------------- /gpfslocalsup/pub/modules-idris-env4/modulefiles/linux-rhel8-skylake_avx512 ------------- openmpi/3.1.4-cuda openmpi/3.1.6-cuda openmpi/4.0.2-cuda openmpi/4.0.4-cuda $ module load openmpi/4.0.4-cuda
$ mpifort source.f90 $ mpicc source.c $ mpic++ source.C
Aucune option particulière n'est nécessaire pour la compilation, vous pouvez vous référer à la rubrique Compilation GPU de l'index pour plus d'information sur la compilation des codes utilisant les GPU.
Adaptation du code
L'utilisation de la fonctionnalité MPI CUDA-aware GPUDirect sur Jean Zay impose de respecter un ordre d'initialisation bien précis pour CUDA ou OpenACC et MPI dans le code :
- initialisation de CUDA ou OpenACC
- choix du GPU que chaque processus MPI doit utiliser (étape de binding)
- initialisation de MPI.
Attention : si cet ordre d'initialisation n'est pas respecté, l'exécution de votre code risque de planter avec l'erreur suivante :
CUDA failure: cuCtxGetDevice()
11. Exécution
L'interactif et le batch
Vous avez deux modes de travail possibles : en interactif et en batch.
Dans les deux cas, vous devez respecter les limites maximales, en temps elapsed (ou d’horloge), en mémoire, en nombre de processeurs et/ou en nombre de GPUs, fixées par l’IDRIS dans le but de mieux gérer les ressources informatiques. Vous trouverez de plus amples informations concernant ces limites en consultant sur notre serveur WEB les pages concernant les partitions Slurm CPU , les partitions Slurm GPU et les pages détaillant les modalités de réservation mémoire pour un travail CPU ou un travail GPU .
Le travail en interactif
A partir de machines déclarées dans les filtres de l'IDRIS, vous disposez d'un accès SSH aux frontales depuis lesquels vous pouvez :
- gérer vos fichiers (création, copie, archivage, sauvegarde, compilation, …) et vos quotas disque au sein des divers espaces disque,
- gérer votre compte (changement de mot de passe, gestion d'un login multi-projets),
- suivre les consommation d'heures de votre ou vos projets,
- soumettre et assurer le suivi de vos calculs sur les nœuds de calcul via le logiciel Slurm,
- vous connecter en SSH aux nœuds de calcul qui sont affectés à l'un de vos travaux afin de surveiller l'exécution de vos calculs,
- exécuter vos codes sur les nœuds de calcul via la commande
srunou via la commandesalloc(allocation/réservation de ressources) ou encore via un shell interactif exécuté sur un nœud de calcul. Pour plus de détails, consultez nos documentations concernant les exécutions interactives CPU et les exécutions interactives GPU.
Remarque : tout code requérant des GPUs ne peut pas être exécuté sur les frontales car elles n'en sont pas équipées.
Le travail en batch
Il existe plusieurs raisons de travailler en mode batch :
- avoir la possibilité de fermer la session interactive après avoir soumis un travail ;
- avoir la possibilité de dépasser les limitations en temps elapsed (ou d’horloge), en mémoire, en nombre de processeurs ou GPUs de l’interactif ;
- faire des calculs avec des ressources dédiées (ces ressources sont réservées pour vous seul) ;
- permettre une meilleure gestion des ressources entre les utilisateurs avec une répartition sur la machine en fonction des ressources demandées ;
- lancer vos travaux de pré/post-traitement sur les nœuds dédiées à grosse mémoire (jean-zay-pp).
À l'IDRIS, nous disposons du logiciel Slurm permettant la gestion des jobs en mode batch sur les nœuds de calcul, sur les nœuds de pré/post-traitement (jean-zay-pp) et les nœuds de visualisation (jean-zay-visu).
Ce gestionnaire de batch contrôle le déroulement des travaux en fonction des ressources demandées (mémoire, temps elapsed ou temps d’horloge, nombre de CPUs, nombre de GPUs,…), du nombre de jobs actifs à un instant donné (nombre total, par utilisateur) et du nombre d'heures consommées par projet.
Il y a 2 étapes essentielles pour travailler en batch : la création puis la soumission du travail (job)
Création d'un travail
Cette étape consiste à écrire dans un fichier toutes les commandes que l’on veut exécuter puis à ajouter, en début de fichier, des directives de soumission Slurm pour définir certains paramètres comme :
- le nom du job (directive
#SBATCH --job-name=...) ; - la limite en temps elapsed pour l’ensemble du job (directive
#SBATCH --time=HH:MM:SS) ; - le nombre de nœuds de calcul (directive
#SBATCH --nodes=...) - le nombre de processus (MPI) par nœud de calcul (directive
#SBATCH --ntasks-per-node=...) - le nombre total de processus (MPI) (directive
#SBATCH --ntasks=...) - le nombre de threads OpenMP par processus (directive
#SBATCH --cpus-per-task=...) ; - le nombre de GPUs pour les jobs utilisant des GPUs (directive
#SBATCH --gres=gpu:...).
Une fois les directives de soumission définies, il est recommandé d'entrer les commandes dans l'ordre suivant ;
- se placer dans le répertoire d'exécution WORK, SCRATCH ou JOBSCRATCH (pour plus d'information, voir notre documentation sur les espaces disque) ;
- copier les fichiers d’entrée nécessaires à l’exécution ;
- lancer l'exécution (via la commande
srunpour les codes MPI, hybrides ou multi-GPU) ; - éventuellement copier les fichiers de résultats que vous souhaitez conserver si vous avez utilisé le SCRATCH ou le JOBSCRATCH.
Remarques :
- La directive Slurm
#SBATCH --cpus-per-task=...fixant le nombre de threads par processus, permet également de fixer la quantité de mémoire disponible par processus. Pour de plus amples informations, veuillez consulter nos documentations sur l'allocation mémoire d'un travail CPU et/ou d'un travail GPU. - Des exemples détaillés de travaux sont disponibles sur notre site Web dans les sections intitulées “Exécution/Contrôle d'un code CPU” et “Exécution/Contrôle d'un code GPU”.
Soumission d'un travail
Pour soumettre un travail en batch (ici le script Slurm mon_job), vous devez utiliser la commande suivante :
$ sbatch mon_job
Votre travail sera placé dans une partition en fonction des valeurs demandées dans les directives Slurm. Nous vous conseillons de positionner au plus juste les paramètres concernant le nombre de CPUs/GPUs et le temps elapsed afin d’avoir un retour de job le plus rapide possible.
Remarques :
- Pour le suivi et la gestion de vos travaux batch, vous devez utiliser les commandes Slurm.
- Le mode batch ne permet pas à l’utilisateur d’intervenir au cours de l'exécution des commandes, sauf pour interrompre le déroulement du job. Par conséquent, les transferts de fichiers doivent s'effectuer sans avoir à taper de mot de passe.
- Les nœuds de calcul n'ont aucun accès à Internet ce qui interdit tout téléchargement (dépôts Git, installation Python/Conda, …) depuis ces nœuds. Si besoin, ces téléchargements peuvent être effectués depuis les frontales ou depuis les nœuds de pré / post-traitement soit avant l'exécution du code, soit via la soumission de travaux en cascade.
- Si vous voulez exécuter un travail sur la machine pré / post-traitement (jean-zay-pp), vous devez utiliser la directive Slurm ci-dessous dans le script de soumission :
#SBATCH --partition=prepost
En l'absence de cette directive de soumission, le travail va s'exécuter dans la partition par défaut donc sur les nœuds de calcul.
Pour tout problème, vous pouvez contacter le Support aux Utilisateurs de l’IDRIS.
12. Formations proposées à l'IDRIS
Formations proposées à l'IDRIS
L’IDRIS dispense des formations à destination aussi bien de ses utilisateurs que de ceux du calcul scientifique en général. La plupart de ces formations figurent au catalogue de CNRS Formation Entreprises, ce qui les rend accessibles à tous les utilisateurs du calcul scientifique, appartenant aussi bien au monde académique qu'industriel.
Ces formations sont principalement orientées vers les méthodes de programmation parallèles : MPI, OpenMP et hybride MPI / OpenMP, clés de voûte pour l’utilisation des supercalculateurs d’aujourd’hui. Depuis 2021, des formations concernant l'intelligence artificielles sont proposées.
Le catalogue des formations proposées par l'IDRIS, régulièrement remis à jour, est disponible ici : liste des formations au catalogue de l'IDRIS.
Si vous souhaitez obtenir davantage d’informations, n’hésitez pas à nous contacter à l’adresse mail : e-cours@idris.fr
Ces formations sont gratuites si votre employeur est soit le CNRS soit une université française. L'inscription se fait alors directement sur le site web des formations de l'IDRIS. Sinon, contactez CNRS Formation Entreprises pour vous inscrire.
Les supports des formations dispensées à l'IDRIS sont disponibles en ligne ici
13. Documentation de l'IDRIS
- Le site Web : l’IDRIS maintient à jour un site Web : www.idris.fr regroupant l’ensemble de la documentation technique (nouvelles du centre, fonctionnement des machines, etc.).
- Les documentations constructeurs : des documents, fournis par les constructeurs, sont accessibles sur le site Web de l’IDRIS. Ils permettent, par exemple, d’avoir une documentation complète sur les compilateurs (Fortran, C et C++), les bibliothèques scientifiques, les bibliothèques de passage de messages (MPI), etc.
- Les manuels : tous les manuels UNIX sont disponibles en ligne grâce à la commande
manexécutée à partir de votre login sur tous les calculateurs de l'IDRIS.
14. Le support aux utilisateurs
La permanence de l'IDRIS
Questions et/ou problèmes techniques
Pour toute question, demande d'information ou problème sur les machines de l'IDRIS, contactez la permanence de l’équipe Support aux utilisateurs. Cette permanence est assurée conjointement par les membres de l’équipe Support utilisateurs HPC (SU-HPC) et les membres de l'équipe Support utilisateurs IA (SU-IA).
Vous pouvez soumettre vos questions :
- par téléphone au 01 69 35 85 55, ou
- par courriel adressé à
Cette permanence est assurée sans interruption :
- du lundi au jeudi de 9h à 18h
- et le vendredi de 9h à 17h30
Note : En période de vacances scolaires (par exemple : Noël, vacances d'été), les horaires peuvent être réduits :
du lundi au vendredi de 9h à 12h et de 13h30 à 17h30.
En dehors des heures ouvrées, les horaires en cours sont indiqués dans le message du répondeur téléphonique de la permanence.
Gestion administrative des utilisateurs de l'IDRIS
Pour tout problème de mot de passe, d'ouverture de compte, d'autorisation d’accès, ou pour nous envoyer les formulaires d'ouverture ou de gestion de compte, vous devez envoyer un courriel à :