Parallélisme hybride (modèle et données)
La méthodologie présentée dans cette page montre comment adapter les dataloaders, les modèles et la boucle d’entraînement pour profiter de plusieurs GPUs pouvant être répartis sur plusieurs nœuds.
Nous nous appuyons sur les pages Parallélisme de données et Parallélisme de modèle, qu’il convient de consulter préalablement.
L’orchestration entre la distribution de modèle et le parallélisme de données ajoute quelques étapes supplémentaires aux documentations précédentes :
- configuration de l’environnement Slurm
- initialisation du code
- adaptations au niveau du modèle
La création des dataloaders est à reprendre sur la page Parallélisme de données. La création du modèle et l’implémentation de la boucle d’entraînement sont à copier de la page Parallélisme de modèle.
Vous trouverez le code complet ici ou sur Jean-Zay dans le répertoire DSDIR. Pour le copier dans votre espace $WORK :
cp $DSDIR/examples_IA/Torch_parallel/bench_DPP_transformer.py $WORK
Ce code est fonctionnel à partir du module pytorch-gpu/py3/1.10.0.
La figure suivante montre un exemple de parallélisme hybride (4 GPUs avec une copie du modèle répartie sur 2 GPUs) :
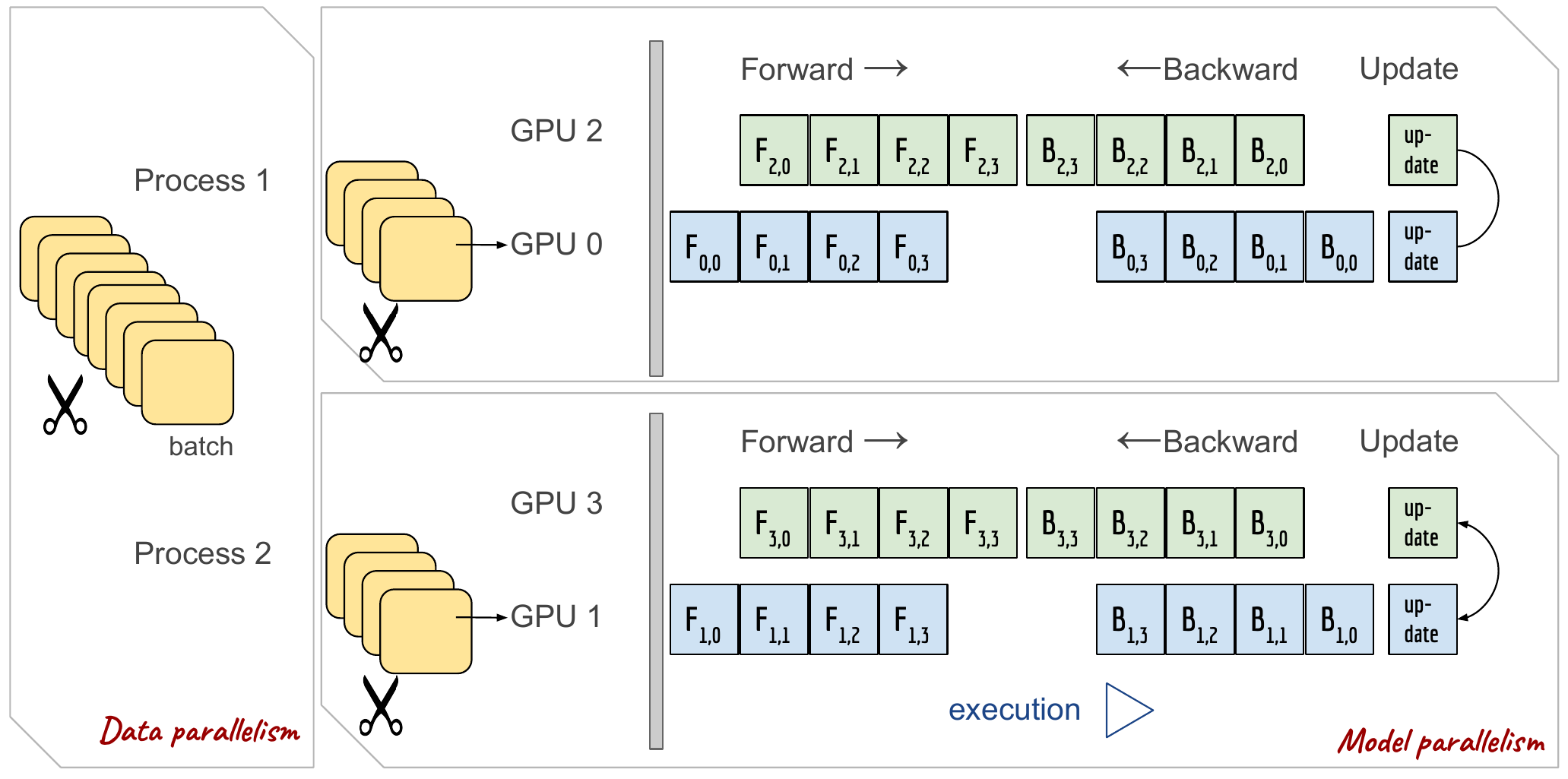
Parallélisme hybride
Configuration de l’environnement Slurm
Pour rappel :
- dans le cas de la parallélisation de données (variante Data Distribution Parallel), il faut réserver autant de tâches par nœud que de GPUs, c’est
torch.distributed.init_process_groupqui se charge d’orchestrer les différentes tâches. - dans le cas de la distribution de modèle, il faut au contraire réserver une seule tâche par modèle et autant de GPUs que nécessaires pour déployer une instance du modèle (dans cette documentation, nous parallélisons un modèle sur deux GPUs).
Il faut maintenant faire attention à créer un nombre de tâches correspondant au nombre d’entraînements à réaliser en parallèle et non pas au nombre de GPUs.
Si le modèle entraîné peut tenir sur 2 GPUs avec le batch size désiré et que l'on veut utiliser trois nœuds comportant chacun 4 GPUs pour l’entraînement, on devra définir les options slurm suivantes :
#SBATCH --gres=gpu:4 # reserve 4 GPUs par nœud
#SBATCH --nodes=3 # reserve 3 nœuds
#SBATCH --ntasks-per-node=2 # cree 2 processus par nœud (donc 2 GPUs par processus)
Cela correspond à 6 processus au total tournant en parallèle et ayant chacun 2 GPUs dédiés.
Et dans le cas de deux nœuds comportant chacun 8 GPUs, la configuration pour 8 processus ayant chacun 2 GPUs dédies sera :
#SBATCH --gres=gpu:8 # reserve 8 GPUs par nœud
#SBATCH --nodes=2 # reserve 2 nœuds
#SBATCH --ntasks-per-node=4 # cree 4 processus par nœud (donc 2 GPUs par processus)
Initialisation du code
Il est important de bien définir les GPUs sur lesquels seront envoyés les entrées du modèle (parallélisme de données) ainsi que les couples de GPUs sur lesquels le modèle sera distribué. Pour cela, on importe le script idr_torch dans le code principal, cela nous permet d'accéder facilement aux variables définies par Slurm (script proposé par l'IDRIS, voir Parallélisme de données).
Récupération des variables nécessaires
Grâce à idr_torch, nous avons accès à diverses informations telles que :
-
hostnames: la liste des noms des nœuds impliqués dans le job. -
size: équivalent de SLURM_NTASKS (nodesxntasks_per_node) qui sert lors de la configuration des dataloaders (num_replicadans le jargon pytorch) -
rank: équivalent de SLURM_PROCID qui permet d’identifier un processus -
local_rank: équivalent de SLURM_LOCALID qui permet d’identifier un processus sur un nœud, et dans notre cas à affecter les GPUs aux bons processus -
cpus_per_task: équivalent à SLURM_CPUS_PER_TASK qui permet de fixer un nombre adéquat de threads pour lire les données (num_workersdans la déclaration du data_loaders).AttentionAttention, la valeur optimale pour
num_workersest rarementcpus_per_taskqui constitue uniquement une indication de la valeur maximale raisonnable
Ainsi que les deux variables suivantes qui sont indispensables car elles servent à créer des listes de GPUs pour la distribution de modèle :
- NTASKS_PER_NODE, variable d’environnement Slurm, accessible uniquement si l’on utilise l’instruction
#SBATCH --ntasks-per-node=xou calculable avec les variables fournies paridr_torch. torch.cuda.device_count(), nombre de cartes GPU visibles du processus (soit toutes les cartes du nœud).
Mise en place des modes de communication
import torch.distributed as dist
# Initialise la communication pour la distribution des données (DDP)
dist.init_process_group(backend='nccl', init_method='env://',
world_size=idr_torch.size, rank=idr_torch.rank)
# Initialise la communication pour le Pipeline Parallelism
dist.rpc.init_rpc(name="worker", rank=0, world_size=1,
rpc_backend_options=dist.rpc.TensorPipeRpcBackendOptions(
init_method="file://{}".format(tmpfile.name),
# Specifying _transports and _channels is a workaround and we no longer
# will have to specify _transports and _channels for PyTorch
# versions >= 1.8.1 (Not True for Jean Zay)
# With Jean Zay, _transports must be equal to ["shm", "uv"] and not ["ibv", "uv"] (like in pytorch doc)
_transports=["shm", "uv"], _channels=["cuda_ipc", "cuda_basic"],
)
)
Adaptations au niveau du modèle
Définition des groupes de GPUs par processus
L’objectif est d’affecter un nombre de GPUs correspondant au nombre de parties du modèle découpé (nombre de séquences) à chaque processus, et à les donner en paramètres lors de la création du modèle :
module_list = []
# En combien de parties le modèle va être découpé
nb_part = torch.cuda.device_count()//int(os.environ['SLURM_NTASKS_PER_NODE'])
# Le numéro du GPU où la première partie du modèle ira
first_part = idr_torch.local_rank*nb_part
# Le numéro du GPU où la dernière partie du modèle ira
last_part = first_part+nb_part-1
partition_len = max((args.nlayers / nb_part), 1)
# L'encoder va toujours dans le premier GPU avec les Transformers
tmp_list = [Encoder(args.ntokens, args.d_model, args.dropout).to(first_part)]
# Ajoute tous les blocs de Transformer dans le modèle et les places dans le GPU approprié
for i in range(args.nlayers):
transformer_block = Layer(args.d_model, args.n_head, args.d_hid, args.dropout)
if i != 0 and i % (partition_len) == 0:
module_list.append(nn.Sequential(*tmp_list))
tmp_list = []
device = int(i // (partition_len))
tmp_list.append(transformer_block.to(first_part+device))
# La tête du modèle va toujours dans le dernier GPU avec les Transformers
tmp_list.append(Decoder(args.d_model, args.d_hid).to(last_part))
module_list.append(nn.Sequential(*tmp_list))
# l'option checkpoint de Pipe doit être configurée sur "never" pour pouvoir marcher avec le DDP
# le modèle utilisera donc beaucoup plus de mémoire que lorsque l'on utilise le gradient checkpointing
model = Pipe(nn.Sequential(*module_list), chunks = args.chunks, checkpoint="never")
Encapsulage du modèle
Après la création du modèle distribué, on rajoute l’instruction suivante, pour le rendre compatible avec le parallélisme de données :
ddp_mp_model = DistributedDataParallel(model)
Cela permet à PyTorch d’envoyer les fractions de batches aux différentes instances du modèle et d’assurer la synchronisation lors de sa mise à jour.
Si l’on regarde le profil de la fonction DistributedDataParallel(...), on y trouve les options suivantes :
device_ids= [...]output_device= i
Dans le cas d’un modèle en parallélisme hybride, il est primordial de ne pas fixer ces variables.
Sauvegarde et chargement du modèle
La sauvegarde des paramètres du modèle se fait uniquement sur le processus maître :
if idr_torch.rank == 0
torch.save(model.module.state_dict(), name)
Le fait d’ajouter le parallélisme de données sur un modèle réparti sur deux GPUs (ou plus), ajoute une “indirection” au dictionnaire de paramètres (model.module au lieu de model), que l’on peut éviter dans le fichier de sauvegarde en récupérant directement le dictionnaire au niveau de model.module plutôt que model (comportement par défaut).
Le chargement doit être réalisé par tous les processus. A noter que nous n’avons testé que des configurations “statiques” : chargement d’un modèle bi-GPU sur une configuration aussi bi-GPU. Pour plus d’informations, se référer au tutoriel Saving and Loading Models.
if load_model:
model.load_state_dict(torch.load(model_path))
ddp_mp_model = DistributedDataParallel(model)
Benchmark
Un benchmark a été réalisé pour comparer les différents paramètres du Pipeline Parallelism et du Data Parallelism (distribution de données).
Les tests ont été réalisés sur 4 et 8 GPUs, avec différents nombres de processus (ntasks). Le nombre de processus va de 1 à 4 pour les tests sur 4 GPUs et de 2 à 8 pour les tests sur 8 GPUs. Lorsqu'il n'y a qu'un seul processus cela veut dire qu'il n'y a que du parallélisme de modèle qui est appliqué et lorsqu'il y a autant de processus que de GPUs cela veut dire qu'il n'y a que du parallélisme de données qui est appliqué.
Le modèle utilisé est un Transformer qui est entraîné sur le dataset IMDB Review dans le but de mesurer les performances de l’entraînement (utiliser un Transformer pour une tâche si peu complexe est déconseillé). Différents batch sizes par GPUs ont aussi été testés, de 32 à 256, et combinés avec différents chunks, de 1 (parallélisme naïf) à 32.
Lorsqu'une donnée est manquante, c'est que la configuration n'est pas possible (par exemple quand on veut faire du parallélisme de modèle alors qu'il y a autant de processus que de GPUs). Les résultats sont les suivants :
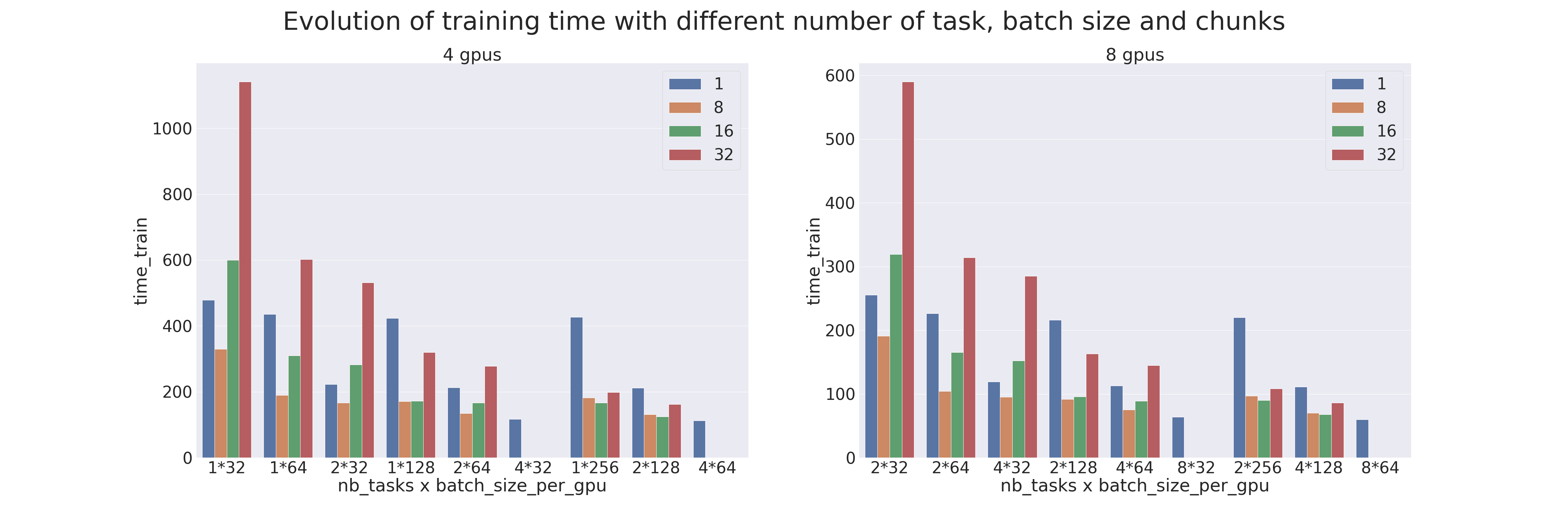
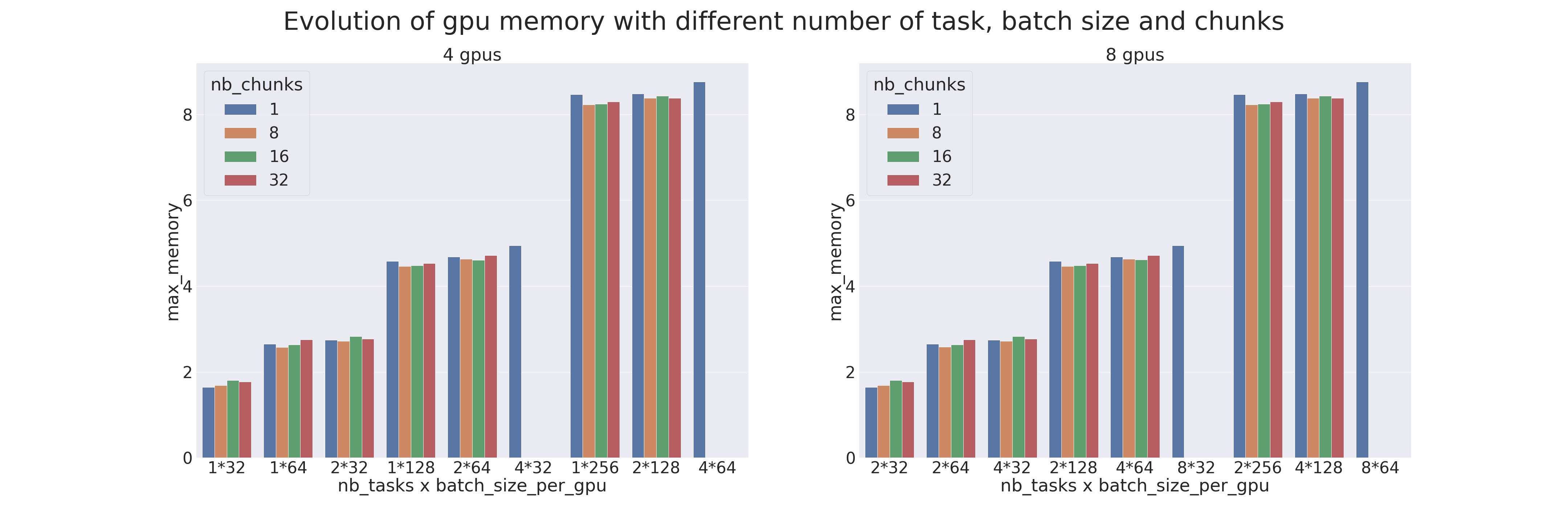
Résultats du bench
Compléments d’information
Quelques remarques pour finir :
- l’optimisation d’un code distribué, modèle et données, est complexe.
- d’autres techniques permettent de limiter la mémoire GPU nécessaire pour un modèle, elles peuvent être plus simples à mettre en œuvre et tout aussi efficaces. Par exemple, la Mixed-Precision (ou AMP), le Gradient Checkpointing, l'optimisation ZeRO.